

 分享
分享

扫一扫手机观看
今天演讲的内容主要分为三部分,支气管扩张(以下简称为支扩)和遗传性支扩的概述,和大家分享5个病例,再重点介绍2种相对常见的遗传性支扩,包括囊性纤维化(CF)和原发性纤毛运动障碍(PCD)。
中国的支扩病因
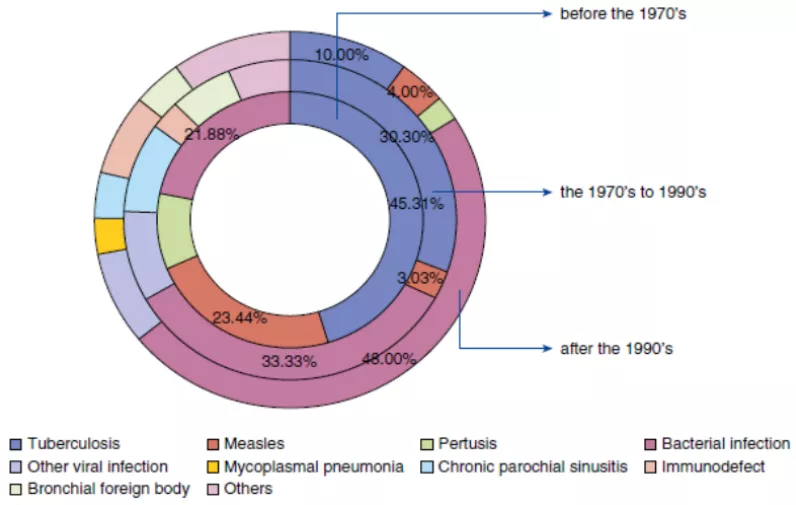
这是2016年徐金富教授团队在《Annals of the American Thoracic Society》杂志发表的关于中国支扩进展的文章,其中把这些年的研究进行了区分,年代不同病因不同,发现在70年代以前,支扩的原因主要是以结核为主导(45%);90年代后,结核的比例下降了10%,细菌感染的比例有所上升(22%→48%),另外,还发现了更多的免疫缺陷(0→9%)。这方面可能是真实的疾病谱的变迁,也可能反映了我们对支扩病因认识水平的提高。
北京协和医院支扩病因分布
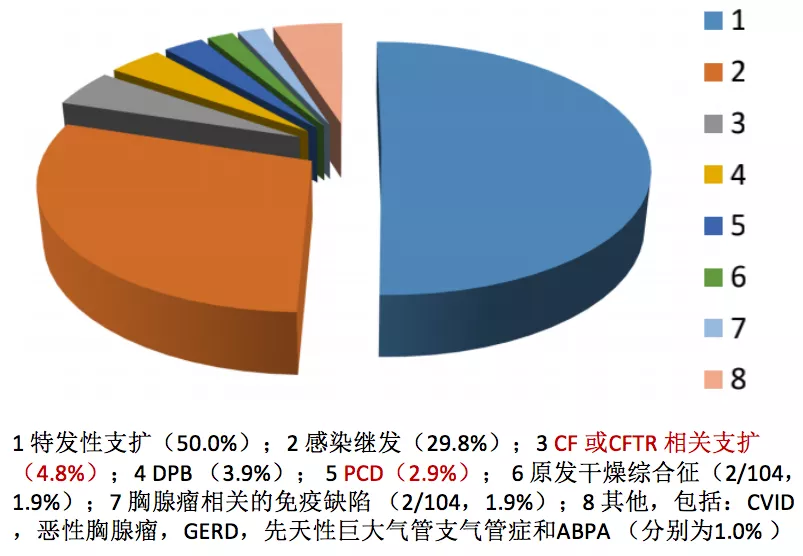
这是2013年总结的北京协和医院支扩的病因。这些病因很复杂,大概有一半未能找到病因,我们把它称为特发性支扩(50%);非常有意思的是有4.8%是CF或CFTR相关支扩,也就是患者符合囊性纤维化或者是找到了一个CFTR基因突变;还有2.9%的PCD,即原发性纤毛运动障碍。在北京协和医院这组的数据中可以看到,CF或CFTR相关支扩加上PCD的比例接近8%,但是这里面存在选择性偏移,因为我们选择的病例观察的数据是来自于北京协和医院的住院病例。相对来说,遗传、罕见的问题容易被收纳进来,实际上真实的数字可能会比这些数据要低很多。但是从这些数据中我们也能够体会到,部分遗传性支气管扩张在支气管扩张的疾病谱中占据一定的比例。
虽然支扩是一种独立的疾病,但是接近半数的支扩病因可以追溯,因此支扩的描述成立之后,进一步区分患者的病因,并寻找针对性的治疗对于患者是非常重要的。
Case 1
这是2006年,我当主治医生时的病例,我几乎在每次讲囊性纤维化的时候都会把这个病例和大家分享。
这是一位16岁的男孩,反复咳嗽、咳痰4年余,加重伴发热3月。入院时有急性感染的过程。胸部CT可见,双肺弥漫性支气管扩张,左肺出现指套征,有片状渗出,是很经典的支气管扩张的病人。
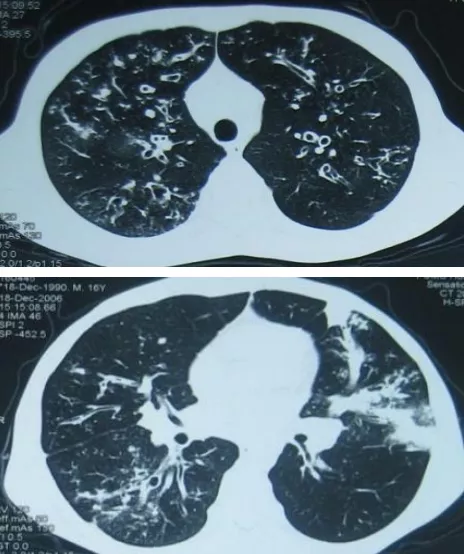
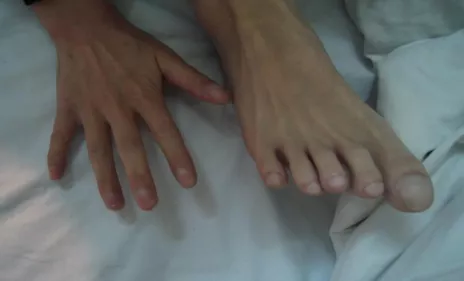
入院后检查发现Serum IgE>5000Ku/L,gm3 (4+),Eos (-)。初步诊断为ABPA。这个男孩有一个特殊的情况,查体发现非常明显的杵状指。我们当时就在思考,一个才16岁的小孩,有多严重的肺部疾病才会出现这么明显的杵状指?
补充病史
我们的住院医生和实习医生非常认真负责,很仔细地询问病史后发现,这个男孩出生5天即患「肺炎」,此后平均每年因「肺炎」住院治疗一次。还有特殊的一点,母乳喂养8个月时出现进食肉类后腹泻,此后长期控制肉食摄入。他之前在北京协和医院的消化科和儿科多次住院,1997年诊断为脂肪泻,予胰酶制剂治疗。否认鼻窦炎、中耳炎病史。
住院医生把支气管扩张和脂肪泻都输入搜索,直接出现了一个叫做cystic fibrosis的疾病。后来他们去询问病人有没有出汗增多,因为囊性纤维化是氯离子通道的异常,经常会出现汗液的异常,发现这个男孩自幼汗液干后皮肤留有盐粒。这样我们考虑他应该是囊性纤维化。
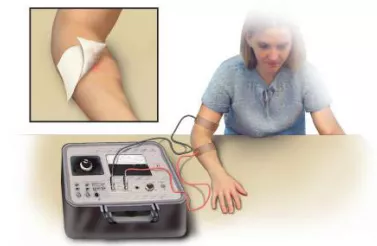
当时是没有检测手段的,我们看到文献上有汗液氯离子检测。后来我们就和住院医生、实习医生一起尝试如何操作,经历了各种各样的困难。由于我们的住院医生是青年男性,作为了正常对照,住院医生的汗液氯离子检测是30mmol/L,病人的检测结果为100mmol/L。最终确诊为 囊性纤维化 。
这就是在日常工作中,通过仔细的病史、查体、多次询问,再结合文献,我们就发现了北京协和医院 首例诊断的囊性纤维化。
囊性纤维化(CF)
简述CF的历史
囊性纤维化是囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)的单一基因突变所致。
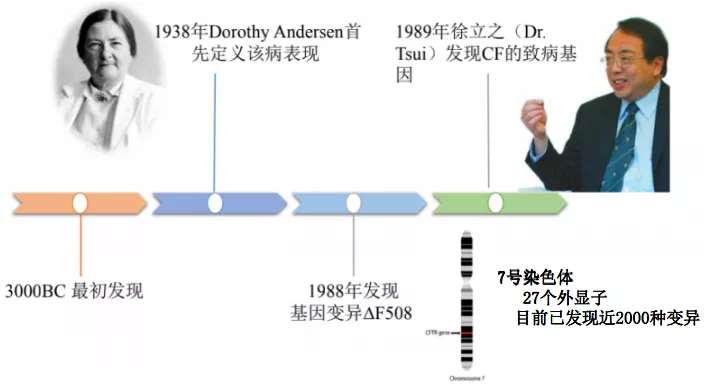
简单介绍一下CF的历史,在3000BC最初发现;1938年Dorothy Andersen首先定义该病表现,把支气管扩张、鼻窦炎、胰腺外分泌功能不全作为此病的特征,另外还有汗液氯离子检测的异常;其实CF真正的基因学的研究时间很短,1988年发现基因变异ΔF508,至今仅有30来年;1989年香港的教授徐立之(Dr. Tsui)发现CF的致病基因,即CFTR,发现在7号染色体第27个外显子异常,目前已发现近2000种变异。
CF在中国是怎么进展的?
1974年,马沛然教授等人进行了我国首例的尸检报道;1995年陈柏华教授等人进行了我国首例的基因分析;北京协和医院在2006年发现首例CF,实际上在这之前北医的李楠医生也曾诊断过;我院于2006年开始尝试做汗液Cl-测定,当时由于技术方法需要摸索,也碰到了很多困难,直到2013年才真正建立好这个体系,开始广泛行汗液Cl离子测定、CFTR测序。随着这些年技术的稳定和改进,我们所做的汗液Cl离子测定和CF的吻合率达到80%以上。
1975年-2006年,全国只诊断了23例CF。但是在2007年-2017年,全国诊断了58例,病例数增加了2倍以上。所以CF病例数是真的逐渐增多,还是我们对这个病的认识增多了,我感觉是后者。
流行病学
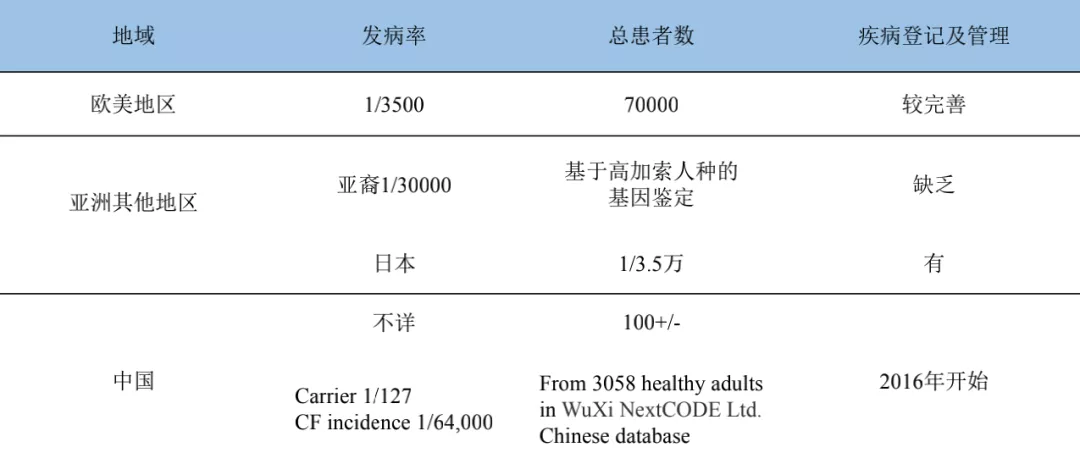
CF在欧美地区的发病率相当高,发病率为1/3500,基因携带率是1/29,也就是说每29个人就有1个人携带了CFTR基因突变,所以他们的总患者数相对较多。对于亚洲地区来说,日本有些研究,总患者数为1/3.5万,这个数字也许能反映咱们真实的状况。
中国的CF数据一直不是很清楚,目前在文献中大概能找到100多例。现在依托于徐凯峰教授和张抒扬书记所领导的中国国家罕见病临床队列研究,遗传性支扩成为该队列研究的一部分,现在登记在册的囊性纤维化患者,北京协和医院是41例,北京儿童医院是39例,加起来共80例,其中不包括已经死亡和拒绝签署知情同意书愿意进行队列随访的病人,所以我国现有的数字应该超过100例。但是想知道我国真实的发病率如何,我们使用无锡NextCODE有限公司的基因检测健康人的中文数据库进行了预测,从3058名健康成人中发现携带率在1/127,按照这样的推测,大概中国人的CF总发生率可能是1/64,000。这样的话,按照中国人口十几亿来说,绝对数字也得有2万多,但是这只是一种推测。
CF的症状
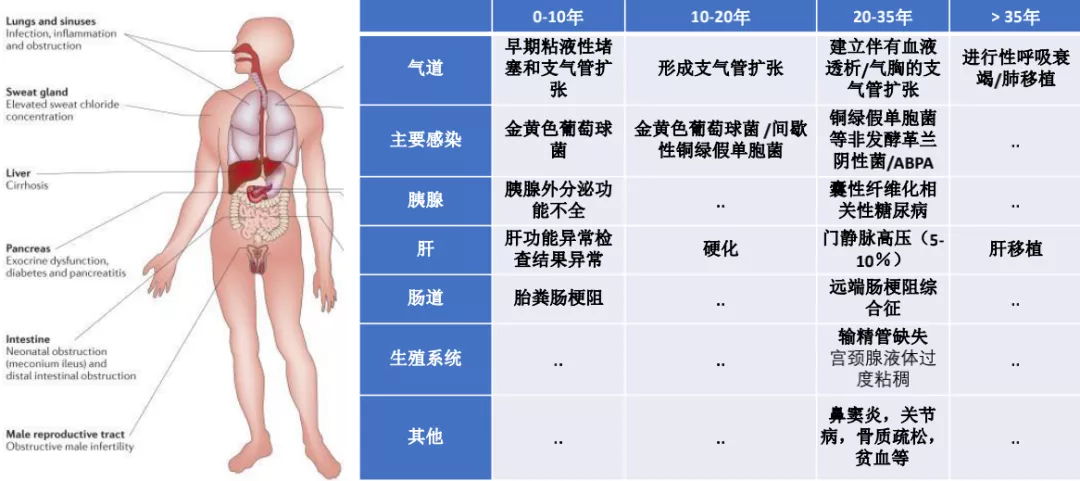
CF的症状与CFTR离子通道的异常在哪些脏器上受累密切相关,主要是呼吸道受累,包括上呼吸道,经常出现鼻窦异常,还会出现反复的支气管扩张和反复的感染。因为胰腺有上皮细胞存在CFTR基因突变,也会形成胰腺外分泌功能不全。由于胰腺外分泌功能不全,导致胰液和胆汁的排泌粘稠度很高,造成一些肠道梗阻,比如胎粪肠梗阻、远端肠梗阻综合征。肝脏也会出现问题,比如肝功能异常等。还有生殖系统的问题,特别是男性,因为有一种常见的疾病叫做先天性输精管缺失,所以很多囊性纤维化的男性如果去做精液测定的话是无精症,这并不是不产生精子,而是由于腺体液体过度粘稠把输精管堵塞或者是发育不良,所以这样的病人如果有生育的愿望,可以做精囊腔穿刺,然后进行体外受精。我们确实也有这样的病人在我院男科的李宏军教授下治疗成功的案例,他专门做CFTR的相关研究。
中国人和欧美人CF的区别
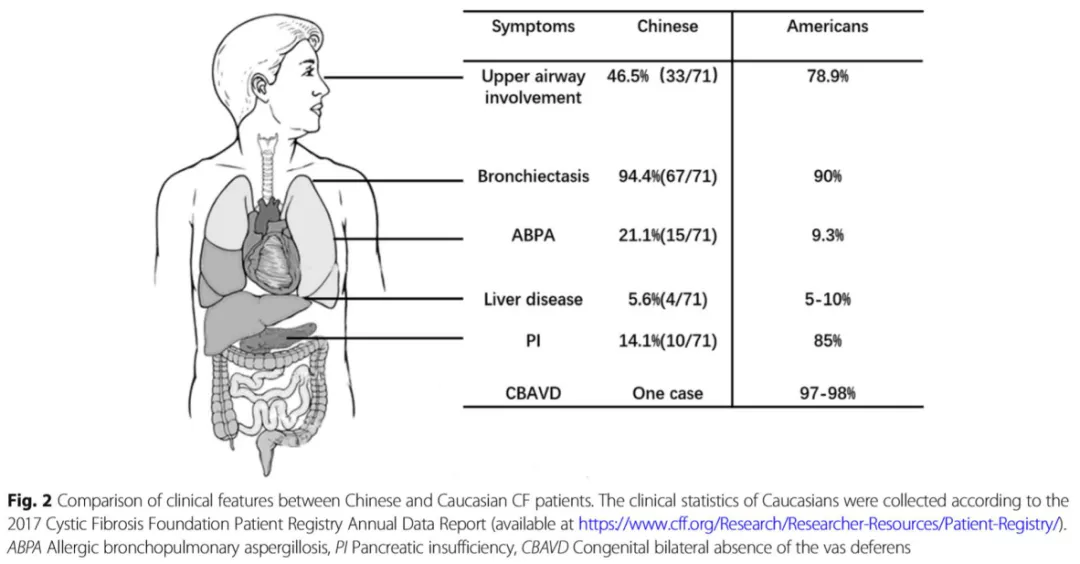
这是2018年我们课题组发布的文章,总结了中国人和欧美人患囊性纤维化的区别。发现中国人出现上呼吸道受累接近50%,欧美人中接近80%;中国人出现支气管扩张和欧美人的数据相似,分别为94.4%和90%;但是ABPA在中国人中比欧美人增加了大概一倍,分别为21.1%和9.3%,我认为可能有些选择性偏移,因为最早的一些CF病人是从北京儿童医院转入我院,他们说这么小就得了ABPA很罕见,让我们帮忙检查有没有什么问题,所以有好几个病人在北京儿童医院没有CF检测手段的时候,我们帮他们确诊下来,但是也不排除是基因异常造成的表型的差异;胰腺外分泌功能不全在中国人中受累是14.1%,在欧美人中是85%,我们看到这个数字很惊讶。因为中国人的胰腺外分泌功能不全往往是通过粪便苏丹Ⅲ染色和询问有无脂肪泻的病史,其他胰腺外分泌功能的检测手段受限,可能造成临床上的漏诊。总体来讲胰腺外分泌功能不全在国外更普遍一些。在我国的71例患者中仅1例成年男性患者,其精液分析符合先天性双侧输精管缺如(CBAVD)。
基因型-人种差别

我们在考虑,既然表型有所不同,那么基因型是不是也有些差别?我们课题组做了相关的研究,病人大概有40例,其实总患者数比这个数字多,因为有些患者已经去世。我们发现在欧美人群中几乎见不到的p.G970D突变,在中国人中最常见(9.8%)。欧美人检测CFTR基因,有70%都是ΔF508突变,而这个突变在中国人中仅有一例,比例非常低。但是还有一个小孩的父亲是德国人,母亲是中国人,所以携带了ΔF508突变。ΔF508突变在欧美人中最常见,而中国人和欧美人的CFTR基因突变差异很大。
CFTR基因异常与表型

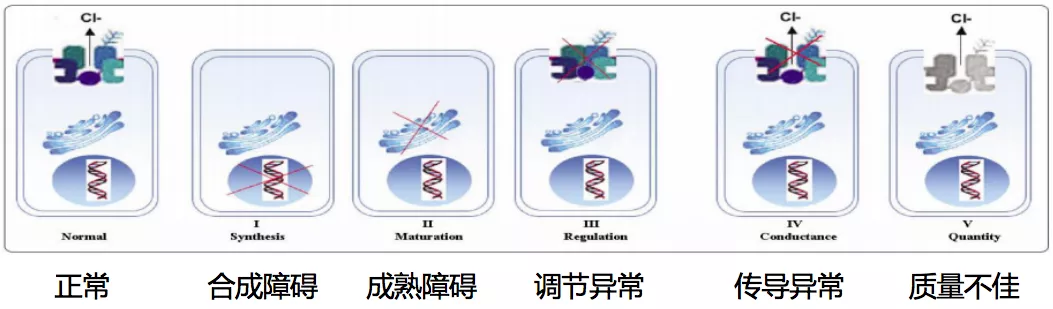
CFTR基因异常与表型有一定的关系,我们目前把CFTR基因异常分为五类。如图所示,合成障碍就是CFTR蛋白质没有产生,这是一类突变;二类突变是成熟障碍,就是不会形成真正有效的蛋白质,二类突变的经典代表就是ΔF508;三类突变是调节异常,跨膜转运的时候运送的调节能力不太好;四类突变是传导异常,五类突变是质量不佳,这两种形式相对来说对总体的蛋白质功能影响比较小。所以我们推测,我们的病人之所以表型上感觉比欧美人群较轻,可能是基因的功能异常主要来自于第四类和第五类突变,前三类突变相对较少。
国外的CF诊断流程
由于发生频率很高,刚才提到欧美的基因携带率是1/29,基本上每个孩子出生后都会先去做婴儿血清免疫反应性胰蛋白酶原(IRT)测定,一旦发现CF婴儿异常,他们就进入基因panel。这个基因panel是基于欧美人常见突变建立的panel,所以在欧美生活的中国人经常检测不到他们想要的突变。
我曾经收到一位美国某大学教授的邮件,他说他领养了一个中国孩子,特别怀疑为CF,但是去了好几家医院都没有确诊,他看到我发表的文章,询问我有没有建议。我告诉他,可能需要做全外显子测序,因为中国人和欧美人的基因突变差异很大。我们之前尝试用欧美的筛选方法,最开始做过基因panel,但是效果都不好,后来现在所有的CF,包括遗传性支扩,我们都直接送去做全外显子测序,这样检测阳性率明显提升。
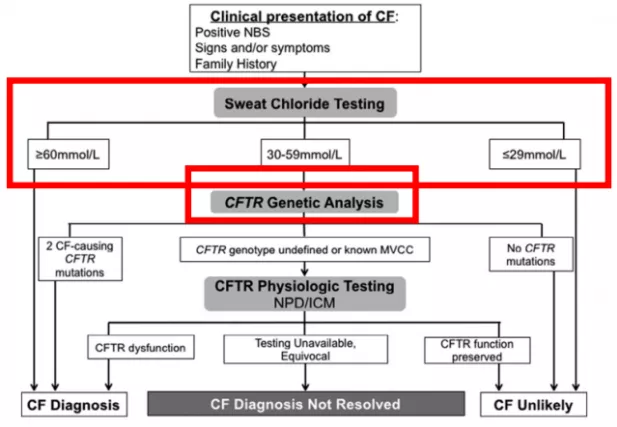
最终CF的诊断还是要依赖汗液氯离子测定,因为这是一项功能检查,只要CF的功能异常,就会导致汗液氯离子的浓度异常,≥60mmol/L:确诊CF;≤29mmol/L:基本排除CF;30-59mmol/L:进一步检验。所以在中国的CF,一般是先进行汗液氯离子测定,一旦发现功能异常,就去进行基因测定。
中国CF的诊断
中国CF的诊断,除了1个或多个器官的临床表现外,还需要有CFTR功能异常的证据:2个部位的汗液氯离子浓度>=60mmol/L。在国外会有阳性家族史,国内目前可能会好一些,因为很多家庭开始有两个孩子了,但是中间中国这么长时间都是独生子女的政策,导致几乎CF患者都没有家族史,或者是偶尔有家族史也是年长的兄弟姐妹去世了,较小的孩子活到现在被确诊。还要满足2个CFTR致病突变。
中国CF的诊断现状:
> 新生儿筛查缺失——不必要/不适用,因为发生率很低,有时候会碰到病人进行遗传咨询,询问既然是CF,是不是要让对方做什么手段能够筛查一下将来不会生出CF的孩子,我认为大概率是没有必要的,因为中国人的CFTR基因携带频率非常低,大概率是找不到的,但是有时候确实是运气不好;
> 汗液氯离子测定及基因检测设施缺乏;
> CFTR基因测序费用高,医保不覆盖,但是现在好在随着二代测序的发展,基因检测的费用已经明显降低;
> 独生子女较多,难获得家族史,这些都造成CF在中国诊断较低。
在临床上碰到哪些情况需要警惕CF?
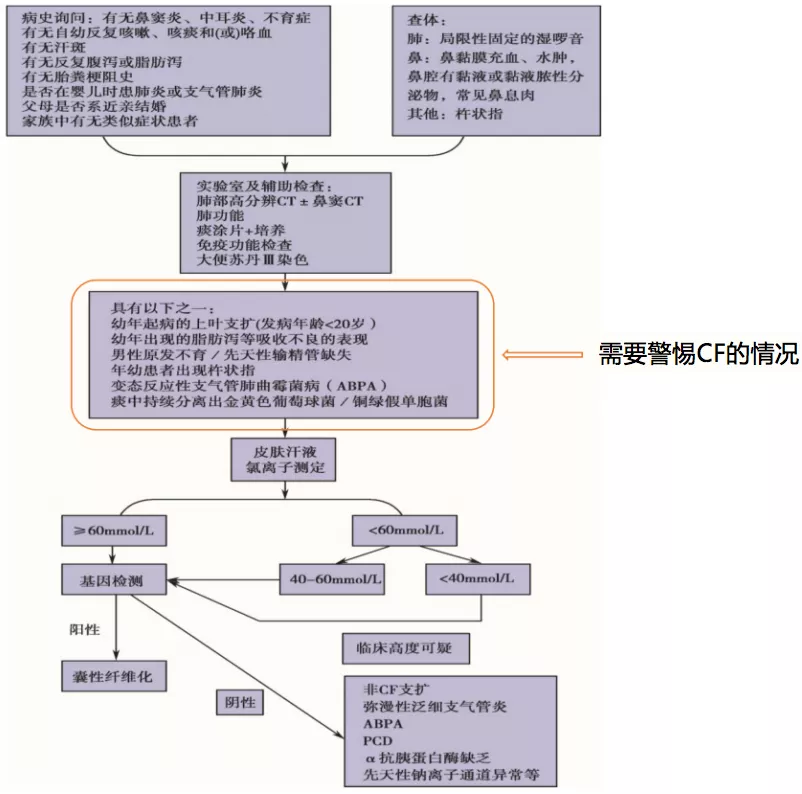
第一,年幼时出现比较明显的支扩,而且这样的支扩往往是双侧对称,而且多数的CF有一个特点是上叶起病,大部分支扩是由于引流不好,重力下垂部位更为多见,但是CF不一样,是由于痰液粘稠造成的管道堵塞,所以CF和平常见到的大部分支扩病变分布不太一样。第二,在幼年出现脂肪泻等吸收不良的表现,有些病人没有脂肪泻,但是生长发育明显比正常同龄儿童迟缓,也要高度怀疑。第三,成年男性如果有原发不育或者先天性输精管缺失,有时候是男科转来的病人。第四,年幼患者出现杵状指,就是刚才提到的Case1,也需要高度怀疑。第五,ABPA,因为中国CF患者有20%左右的ABPA发生率。第六,痰中持续分离出金黄色葡萄球菌/铜绿假单胞菌,都是需要警惕CF的情况,这时我们会建议患者去做汗液检测。
小结
总体而言,CF在中国诊断面临许多困难,临床表现不典型;而且多数医院不能进行汗液检测和基因鉴定,我所知道的目前确定能做汗液测定的是北京协和医院和北京儿童医院,其他医院不太了解;欧美常见CFTR基因检测位点阴性。中国存在不同的CFTR基因谱,影响其功能。因此,建立具有中国特色的基因筛查平台和药物筛选平台迫在眉睫。
CF如何治疗?
我们了解一个遗传病的目的是什么呢?最终还是希望改变患者的预后。
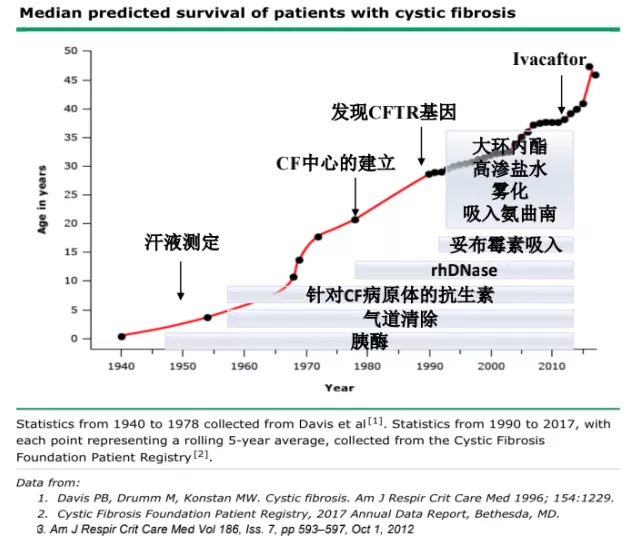
CF在1938年发现的时候,存活率接近0。但是随着近些年的进展,胰酶、气道清除技术、针对CF病原体的抗生素、rhDNase、妥布霉素吸入、雾化抗生素、雾化高渗盐水、大环内酯的使用,仅仅是这些治疗,就使患者的存活时间明显改善,大部分患者能够生存到三四十岁。
后来在2012年有一项重大进展,Ivacaftor是针对G551D基因突变的药物,其实就是三类突变的修复剂,这项治疗明显改善了病人预后。有一项数据,自从Ivacaftor进入临床使用后,现在病人的存活率,尤其在欧美管理比较好的地区,病人平均寿命能够达到60岁以上,已经很接近正常人的寿命了。即便是没有基因靶向治疗,病人的寿命也得到了非常大的提高。怎么才能获得呢?关键就是严格的患者的管理以及患者的自我管理。
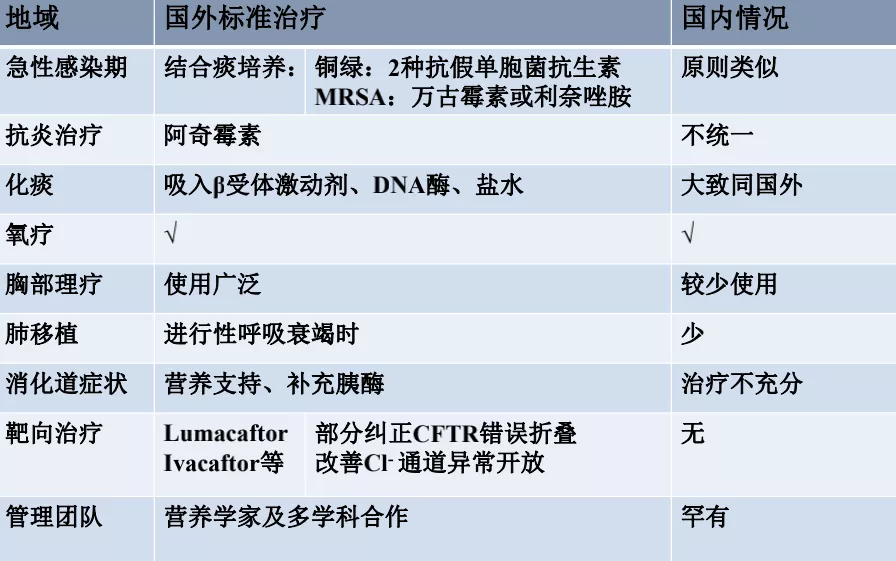
治疗方面,除了急性感染期抗感染治疗以外,目前比较标准的CF治疗方法包括阿奇霉素,化痰药物,氧疗,胸部理疗(非常重要),肺移植,对消化道症状的管理也很重要,因为这类病人经常会出现严重的营养不良,所以胰酶的补充、如何选择营养制剂,包括不能吃普通的油脂,需要用中链氨基酸来替代,这些都会明显改善预后。目前有两种上市的靶向药,但是中国没有,后面我会讲到是否应该使用。还有营养学家及多学科合作,在国内推广不太好。
如何精准治疗?
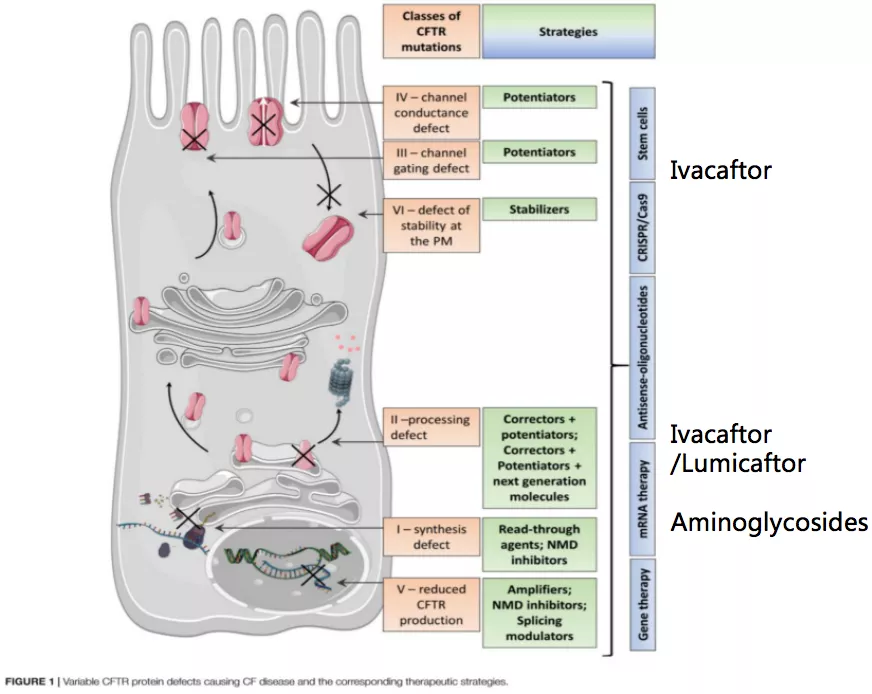
目前已有并且确切对CF精准治疗有效的是Ivacaftor、Lumicaftor、Aminoglycosides(氨基糖苷类药物),这些药物都可以尝试用于患者。但是这两种药如何使用,我先举一个例子。
Potentiators——Ivacaftor (VX-770)
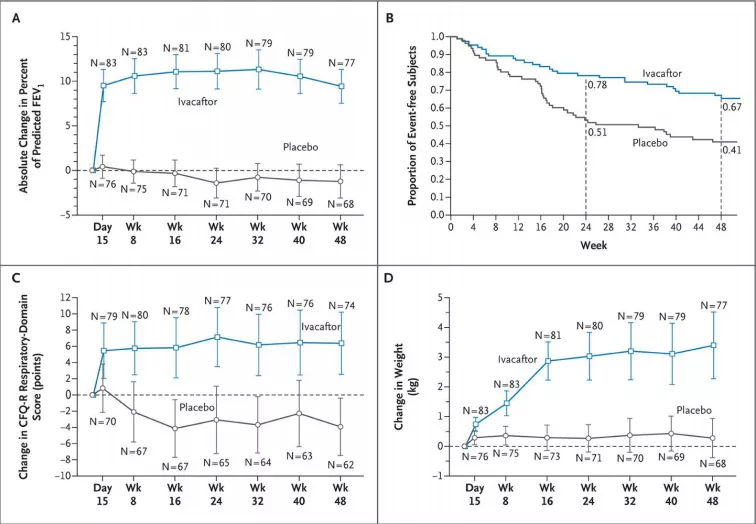
这是《新英格兰医学杂志》发布的一篇文章,Ivacaftor用于G551D(class III)和R117H(class IV),可以增加氯离子流动,也就是精准治疗的典范。其实在那一年奥巴马提出精准治疗的时候,举的例子就是这个药物。在2012年获得FDA批准用于临床,明显改善患者的生活质量,甚至对FEV1的提升也非常明显,能够增加体重,改善肺功能。因为2012年才进入临床使用,真正对患者寿命的影响,至少要在10年或者20年后才能真正看到结果,但是我认为这个药物的前景很好。
中国人有合适的靶向药吗?
发现Ivacaftor以后,尝试了很多Ivacaftor适用的突变靶点的研究,发现目前除了最开始用于临床研究的G551D以外,还可以用于G178R、S549N、S549R、G551S、G1244E、S1251N、S1255P、G1349D、R117H、A455E、E193K、R117C、A1067T、F1052V、R347H、D110E、D110H、F1074L、R352Q、G1069R、R1070Q、D579G、K1060T、R1070W、D1152H、L206W、S945L、D1270N、P67L、S977F、E56K、R74W、711C3A→G, 3272-26A→G, E831X, 2789C5G→A或3849C10 kb C→T等。
但是非常遗憾的是,这些突变在中国现有病人的基因检测谱中都没有发现。虽然现在国际上已经有药物,包括Ivacaftor和Lumicaftor,但是对于F508携带者而言,中国的一例已经去世,还有一例现在在德国生活,可能也用不上这些药物。目前为止,还不知道哪些病人有可能使用这些药物。
目前正在研究的药物有VX-661(Tezacaftor)、VX-659、 VX- 445、VX-440等
国外的CF个体化药物选择
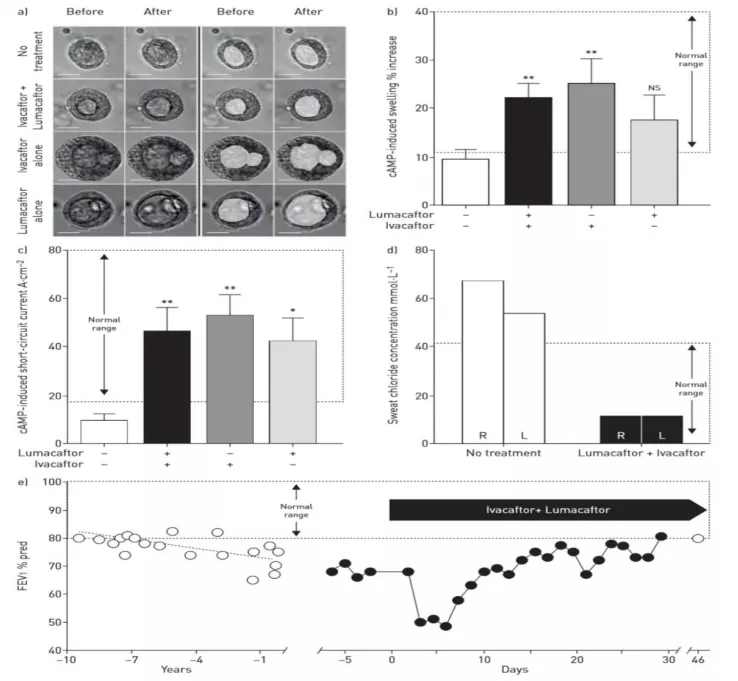
在国外有时候也会碰到一些罕见突变,他们是如何解决的呢?这是在《European Respiratory Journal》发布的一篇文章,这个病人是罕见突变,自己坚持治疗,但是他的突变是不符合这些药物的,强烈要求医生用药,主动提供各种便利,取了一大块鼻粘膜给医生用于细胞培养。非常高兴的是,做了细胞培养,加了药物以后,发现细胞张力能够明显改善。后来这个病人尝试使用Ivacaftor以后,肺功能和汗液氯离子检测都有所改善。因此,建立CFTR细胞模型,药物的筛查可能是精准治疗的必经之路。基于这个想法,我们也在尝试做一些研究。
中国CF患者的出路在哪里?
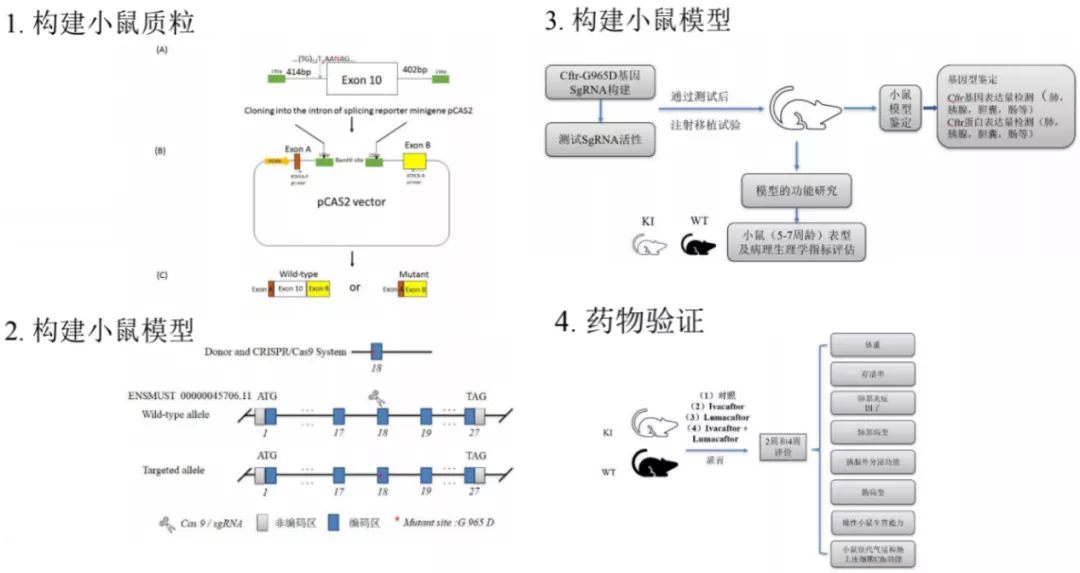
因为我们课题组已经发现G970D是中国CF患者中最常见的突变,虽然有限,但确实有些纯合突变。于是我们尝试构建小鼠质粒,现在小鼠已经孵育出来了,还在培育中。下一步准备进行小鼠的功能鉴定,一旦建立好小鼠模型,我们就可以尝试使用一些药物干预,如果发现G970D用现有的药物有效,那么就可以尝试开始病人的治疗方案。这是我认为中国CF患者的出路,但是遗憾的就是,除了G970D以外,中国人的大部分突变都是非常散发的,因此我们很难获得一大批病人有相同的突变去研发药物。所以,将来治疗罕见病的道路仍然艰难,很难有药厂愿意参与其中,因为获利十分有限。
Case 2
这是一个典型病例。一位29岁的男性,反复咳喘20多年,加重1年。自幼发现内脏转位,鼻炎鼻窦炎史6年。肺功能:FEV1 0.99L 23%,FEV1 /FVC:41%,舒张试验阴性。胸部CT显示内脏转位,支扩,诊断非常明确,Kartagener综合征。
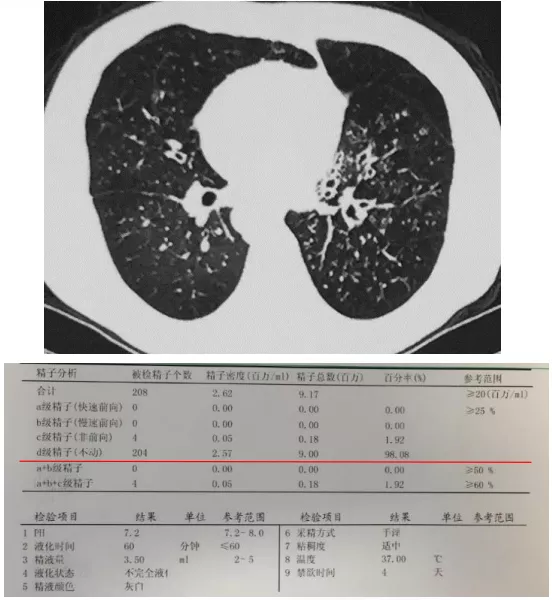
病人已婚未育,父母非近亲,无家族类似患者。我们给他做了精液测定,和CF特别不一样的是,PCD是有精子的,但是精子的活力很差,a+b+c级精子加起来的活力都很低,但是CF常常根本没有精子。
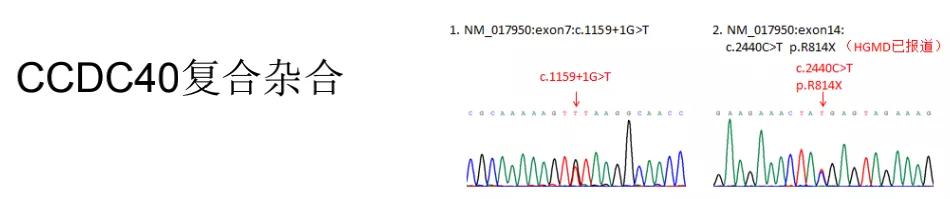
由于现在有了更好的手段,我们就给病人进行了基因检测,发现是CCDC40复合杂合突变。所以这个病人的诊断为PCD。
原发性纤毛运动障碍(PCD)
PCD临床表现
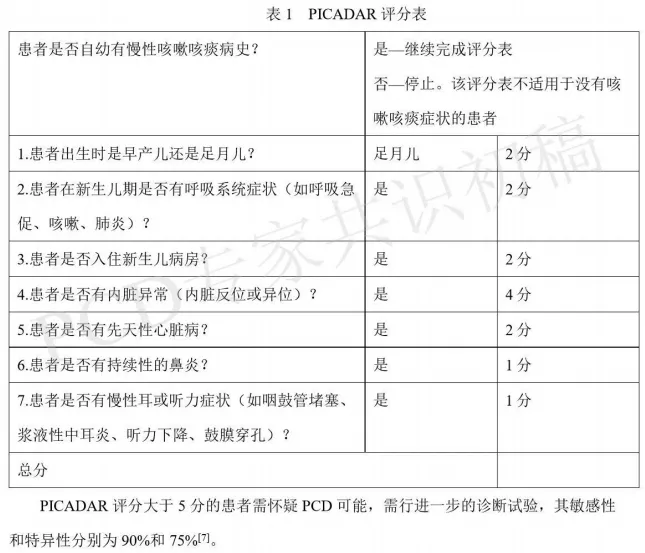
PCD的发生率为1:10.000-20.000。临床表现会出现反复呼吸道感染,特别是儿童期起病者。和CF非常像,也会有鼻窦的症状,很少出现肝硬化,但是也会有胆道的问题出现,因为凡是有纤毛分布的区域,这些细胞如果出现异常,也会出现相应脏器的异常。PCD的症状与哮喘、免疫缺陷和CF难鉴别。欧美国家使用了PICADAR评分表,上图所示表格中的评分越高,倾向于PCD的可能性越大。
需怀疑PCD——但是这些症状可以很轻微或者不出现:
•不能解释的新生儿呼吸窘迫
•早发的鼻充血
•鼻腔分泌物自新生儿期出现并持续终生
•慢性湿性咳嗽
•早发下呼吸道感染
•反复浆液性中耳炎
•内脏转位
•支气管扩张(半数儿童,几乎全部成人)
纤毛结构异常导致的器官病理改变
ERS指南和中国儿科共识对比
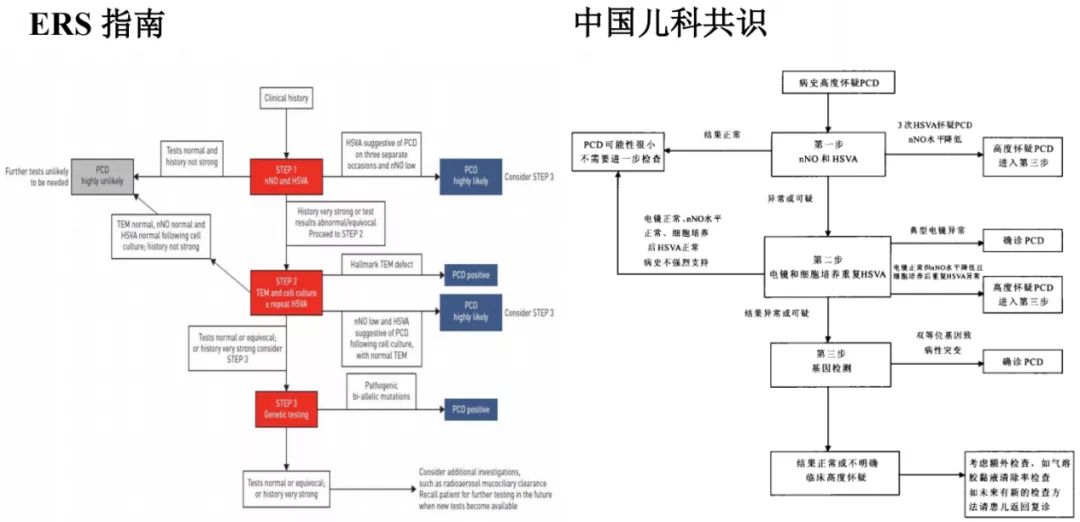
基于这些研究,ERS在2017年发布了共识。根据这个共识,2018年中国儿科也发布了共识,我看到这版共识的内容,中国基本上是仿照了欧美的指南。步骤为:病史高度怀疑PCD,第一步先做nNO和HSVA,下面会讲什么是HSVA,现在北京协和医院能做nNO,但是我知道这个检测受到诸多因素的影响,国际上目前也没有共识表明临界值在哪里,在中国有很大困难。第二步是电镜和细胞培养重复HSVA,第三步才进行基因检测。我个人认为这项共识在中国比较难以实现。
ATS指南
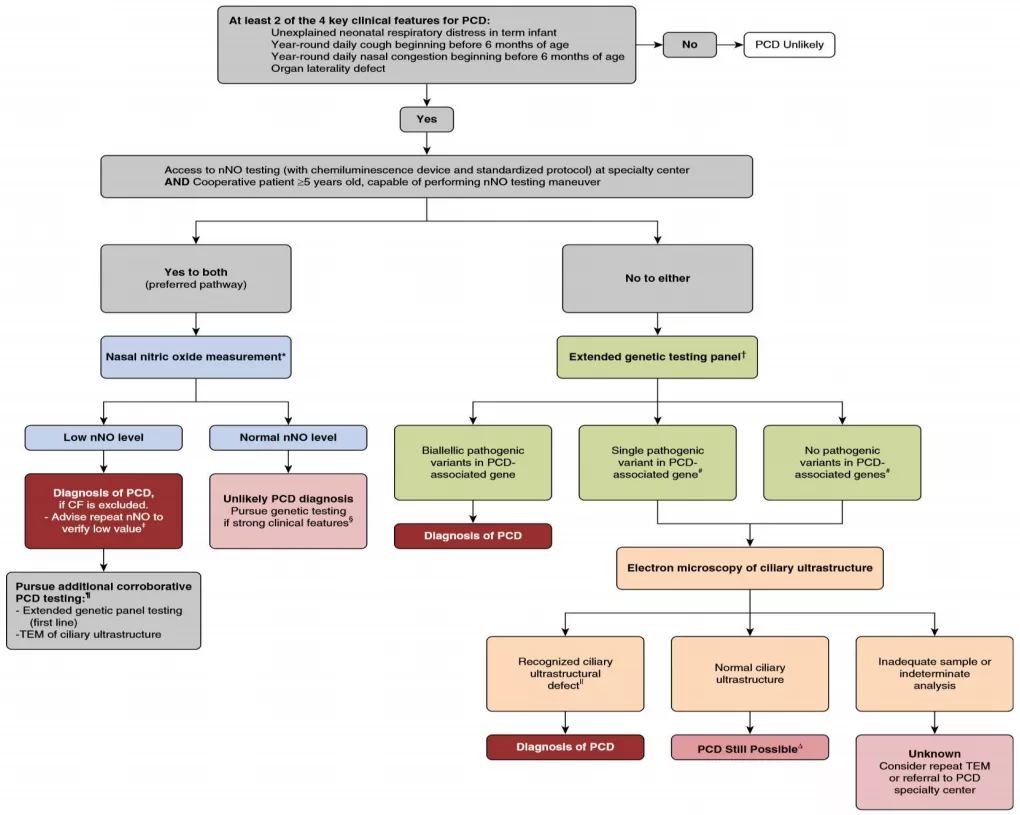
在2018年ATS发布了指南,我个人认为这项指南更符合中国的国情。常见的临床表现,比如呼吸道症状、常年咳嗽咳痰、鼻腔充血或者脏器转位等。这时如果考虑为PCD,除了用nNO测定以外,提到如果没有条件进行nNO,可以直接进入基因测序。
哪些患者应该进行诊断性的检查?

nNO——ERS
大家可以简单看一下nNO检测如何操作。其实就是一根细管,类似呼出气一氧化氮的检测方法,只不过呼出气一氧化氮的检测是用口唇包紧过滤器,nNO是利用鼻塞式探头置于前鼻孔直接采集鼻呼出气。
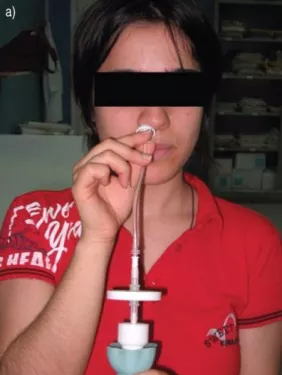

nNO——ATS

纤毛的获取
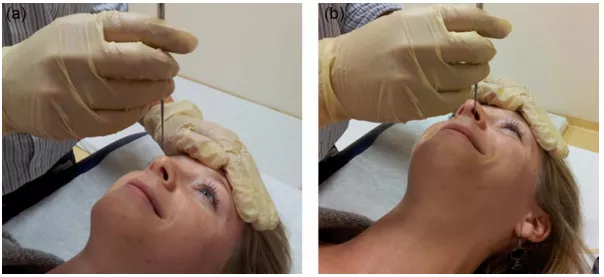
关于电镜,目前我个人比较推荐支气管粘膜,因为鼻粘膜容易造成鼻上皮化生,无法找到纤毛柱状上皮,所以做了也没有意义。纤毛柱状上皮可以通过毛刷、刮器或活检钳获得,后2者可以获得更多标本,并进行纤毛运动的观察,最好用显微镜验证标本是否满意再让患者离开。用于HVMA的标本需放置于缓冲液中,尽早检查;用于TEM的标本用3%的戊二醛固定,可以储存。
Case 3
一个17岁的女孩,自幼反复咳嗽、咯痰,3-4岁后每年发热10次需抗生素治疗。鼻窦炎、中耳炎史,10岁时因右中叶不张行切除术。家族史(-)。外院查CFTR基因杂合突变,c1408G>A, p. V470M杂合,是单基因的杂合子。
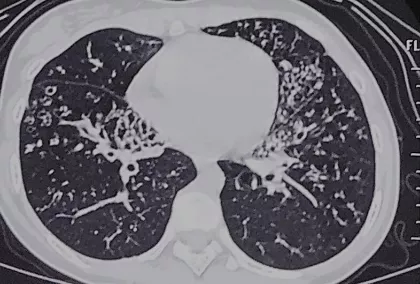
当时女孩的母亲来询问自己的女儿是不是得了囊性纤维化,我观察了CT是以下肺为主的,不符合平常看到的囊性纤维化的改变,更怀疑为PCD。病人也比较配合,于是做了支气管粘膜电镜,但是非常遗憾,突变特征为纤毛形态正常,当时我们没有条件做高频电镜。后来我提出做基因检测,但病人家里的经济条件不是很好没有进行。
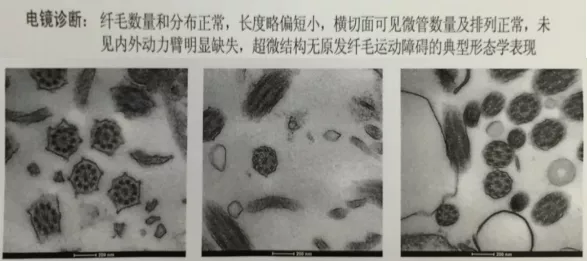
过了好几年,我们自己能做全外显子测序的时候,找到病人做了PCD基因检测,发现2个PCD的杂合突变分别来自父母,突变的特征就是纤毛形态正常,功能异常。
Transmission electron microscopy(TEM)——ATS

透射电镜(Transmission Electron Microscopy,TEM)


PCD可能存在大量的漏诊
因为我们平常看到的Kartagener综合征,鼻窦炎、内脏转位再加上支扩就很容易确定是PCD,像上述Case3中的女孩没有内脏转位,所以对于无内脏转位的病人容易被漏诊。我们2010年总结我国文献中具有内脏转位的PCD所占高达170/178例(95.5%);2016年上海复旦大学附属中山医院2016年总结127例中国的PCD病例,内脏转位的患者仍高达94%。但是真实的内脏转位发生率应该是多少,因为它是从定向转位变成随机转位,应该理论上只有一半人会出现内脏转位,所以这部分没有发生内脏转位的在中国是被大量漏诊的,无内脏转位的诊断率并未提高。
High-speed video microscopy analysis(HSVA),高速录像显微镜——ERS

如果有做HSVA的条件能够确诊是最好的,之前我一直以为国内没有单位能够大规模进行HSVA检测,但是最近我看到了2020年复旦大学附属儿童医院发布的一篇文章,50例PCD病人为一组,大部分都做了HSVA。我知道北京301医院可以进行HSVA检测,我们正在与他们进行合作。所以我认为国内的诊断技术也在明显提高。
HSVA



目前,即便在ATS指南上也不把HSVA作为一线诊断方案,因为受到很多技术条件的限制。
PCD的遗传学诊断
对于遗传方面,如果没有条件做HSVA,也没有条件做nNO,可以直接做基因测序,这是PCD的重要确诊手段,超过30种基因可以覆盖>60%的PCD患者。但目前在我国实施还存在一定困难,因为费用昂贵;独生子女率高,家系获得困难,不易建立遗传家系识别和检测;二代测序不普及,直接测序耗时且阳性率低;PCD为多基因遗传病,我国商业化诊断试剂盒标准化不规范;不断发现新的突变,令现有的测序内容需要不断更新。
PCD临床表型与基因型的关系

现在认为PCD临床表型和基因型存在一定关系。刚才提到的Case2的CCDC40患者,他的FEV1只有20%多,属于上图表格中的严重型,虽然他的年纪只有29岁,但是肺功能恶化非常快。
目前,基因测序现有40余种基因异常,可以解释60-70%的病例,还有30%是基因阴性的,因为这是一种复杂的基因遗传病,不是单基因病,多数为常染色体隐性遗传,少数为X连锁或显性遗传,所以现在能检测到的还是比较困难的。
PCD国内研究进展
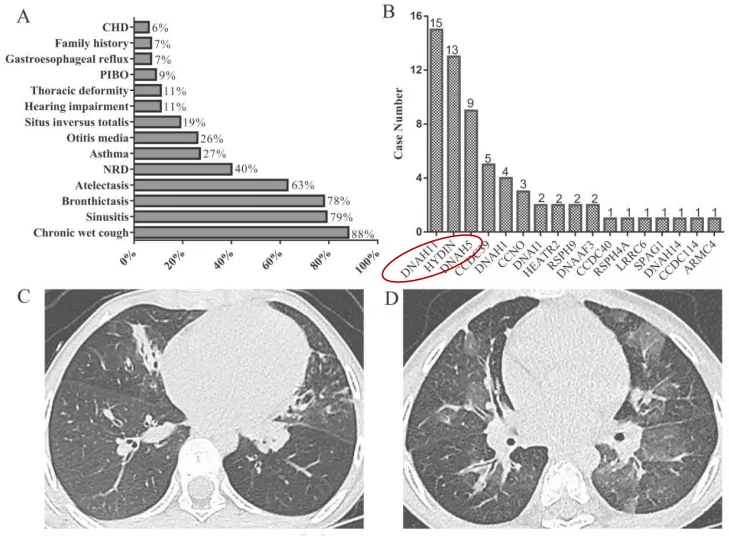
国内近些年对PCD的研究有非常大的进展,包括我们自己课题组也进行了研究,我们的病例数并不是很多,大概30多例。这是北京儿童医院做的报道,发现PCD最常见的突变是DNAH11。北美报道PCD的最常见致病基因变异为DNAH5,其次是DNAH11,DNAI1和CCDC39,他们还发现了HYDIN似乎与内脏转位相关。
Case 4
22岁的男性,晨起后咳嗽、咳黄痰、胸闷憋气,活动耐力减低,登5层楼较前费力。无发热、咯血、胸痛。既往:鼻窦炎3年,08年行「肛周脓肿」手术,间断白细胞下降4余年。家族史(-)。
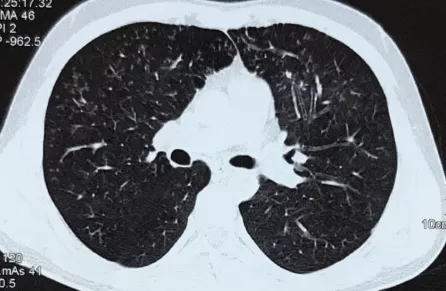
支气管粘膜活检电镜:
上皮细胞表面少量短小纤毛
微管排列基本正常,内动力臂普遍缺失
部分纤毛外动力臂短小、方向异常或缺失
精液常规+质量分析:
a级精子 2.80%
a+b级精子 8.62%
诊断为:原发性纤毛运动障碍。
我们本来以为诊断的很顺利。后来这个病人于2017年在皮肤科看病,发现四肢皮肤疣状增生8年,面积进行性增大,怀疑他得了自身免疫缺陷疾病。于是做了全外测序,一方面符合我们的判断,存在PCD;同时发现了免疫缺陷21型存在,这是一个新的基因。
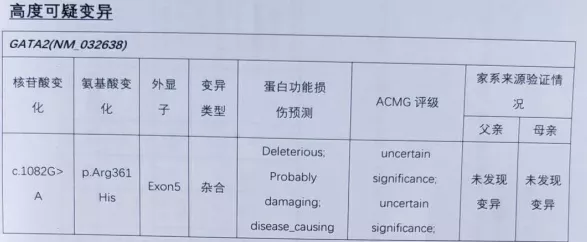
后来我们与北京儿童医院有经验的专家讨论,这到底是因为自身免疫缺陷造成的感染导致的感染后的电镜改变,还是他同时患有PCD和自身免疫缺陷这两种疾病。经过讨论,我们一致认为确实是两病共存。后来北京儿童医院也发现他们那一组共81个病例,有百分之几的病例也是PCD和自身免疫缺陷共患病的情况。所以我认为随着对疾病的认识,我们也在不断地推翻我们自己的想法,再加深认识,一直在跟病人学习。
基因检测——ERS

基因检测——ATS

检查结果不明确
当临床高度怀疑PCD,但是诊断性检查结果模棱两可,TEM正常和接近正常的nNO的时候。临床上可以先按照PCD治疗,因为所有的支扩除了囊性纤维化,非囊性纤维化的支扩治疗的基石都是相似的,都是物理治疗,如果长期定植菌的话可以给予长期大环内酯或者长期抗生素治疗或者雾化生理盐水都是有效的。
Case 5
一位23岁的男性,反复发热、咳嗽咯痰14年,否认家族史。刚开始看到胸部CT,好像上叶支扩,但是仔细看和CF又不同。后来回顾病史发现这个病人有肺脓肿史、自幼反复面部反复脓疱、脓肿史。查痰培养MRSA,血IgE >5000U/L。
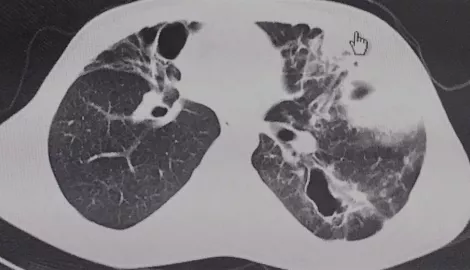
所以我们这时候就发现了另一个常见的引起遗传性支扩的疾病,做了基因测序,发现STAT3基因阳性,诊断为高IgE综合征。
遗传性支扩
支扩的遗传性支扩的诊断路径
遗传性支扩包括了一大组疾病,除了CF、PCD、高IgE综合征,还有钠离子通道异常、Α1抗胰蛋白酶缺乏、X连锁无丙种球蛋白血症、Marfan综合征等。
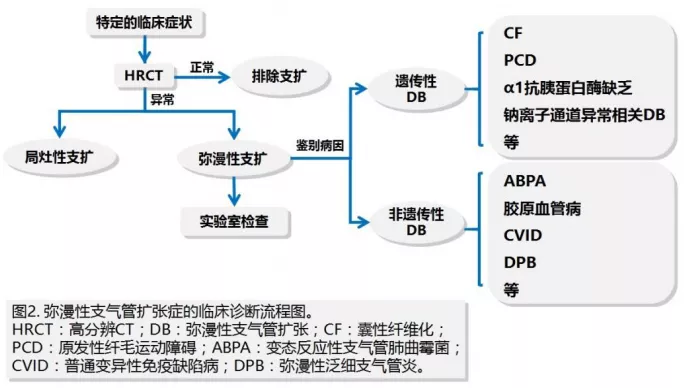
我们在临床尝试建立了一个临床诊断路径,对于弥漫性支气管扩张症来说,先去做一些常见的其他原因的筛查,包括ABPA、胶原血管病、免疫球蛋白缺乏、DPB等,如果排除,会进入怀疑遗传性支扩的诊断筛查流程。
遗传性支扩的基因检测流程
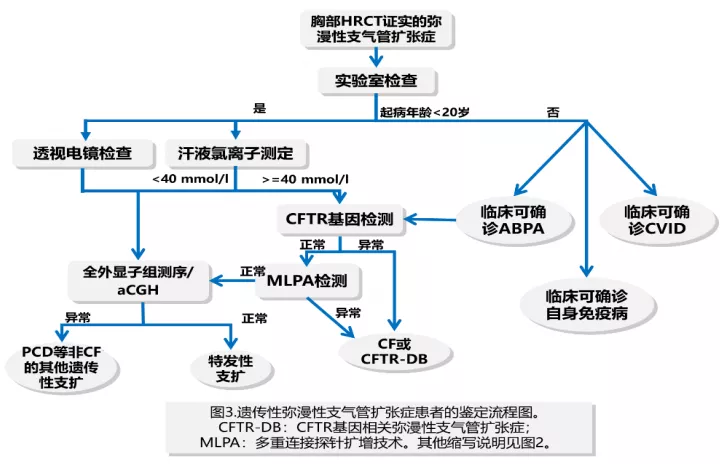
可以先做透视电镜或者汗液氯离子测定,然后再结合基因测序的结果进行判断。这是国外文献的一段话,我觉得说的特别好:支气管扩张只是一个描述性名词,而不是一个诊断,这只是一个诊断的开始,而不是一个诊断的结束。所以对于很多病人,尤其是中国有些病人可能年幼的时候条件受限,没有来过大医院,所以很可能即便是遗传性支扩,就诊的时候并不是幼年的患者,那就要通过非常细致的查体、询问病史获得真实的诊断。
支扩的阶梯治疗
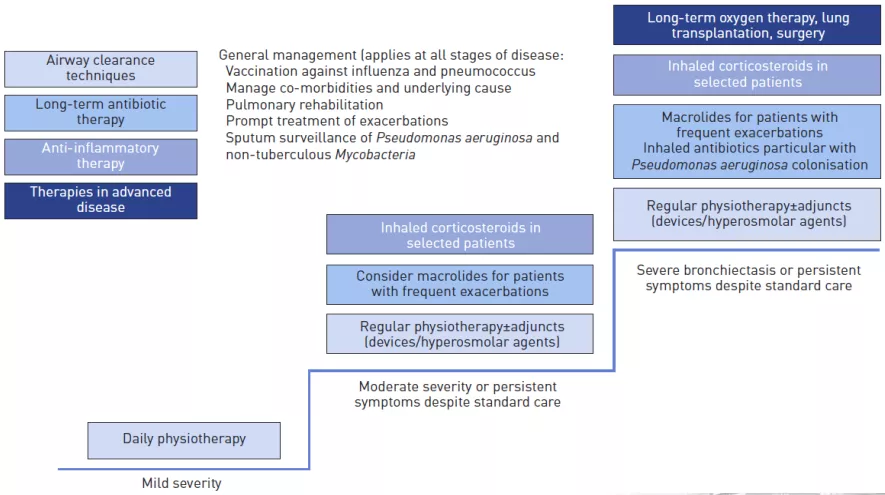
减少症状;改善生活质量;防止与不良结局相关的急性加重。
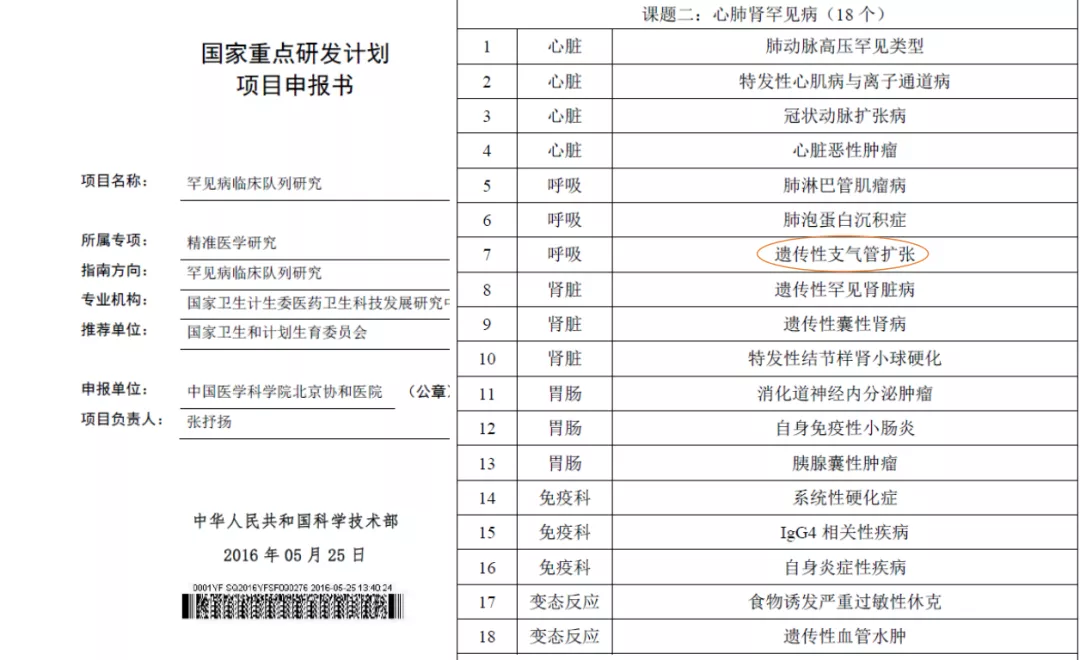
国家越来越重视遗传性支扩的研究,国家十三五的的罕见病队列研究中把遗传性支扩也包括了进来。

这是徐凯峰教授牵头建立的中国罕见病联盟呼吸病学分会成立大会。随着这些研究的进展,我相信遗传性支扩会得到越来越多的关注,也会得到更多的认识。最后感谢一下我们的团队,也感谢在这个过程中所有支持过我们的人,谢谢。
志谢
•医科院基础所遗传系:刘雅萍、黄尚志教授
•北京协和医院呼吸与危重症科:徐凯峰教授、陈珂琪
•哈尔滨医科大学:张学教授
•北京协和医院检验科
•北京儿童医院呼吸科:赵顺英教授
•北京大学附属人民医院电镜室:郑姝颖老师
参考文献
[1]Jie-Lu Lin, et al. Ann Am Thora Soc 2016, 13, 609-616
[2]田欣伦等,中国呼吸和危重监护杂志. 2013,12
[3]Xiaobei Guo, et al. Orphanet J Rare Dis. (2018) 13:224
[4]Elborn JS. Cystic fibrosis[J]. Lancet. 2016 Nov 19; 388(10059): 2519-2531.
[5]Cutting G R. Cystic fibrosis genetics: from molecular understanding to clinical application[J]. Nature Reviews Genetics, 2015, 16(1): 45-56.
[6]Xiaobei Guo, et al. Orphanet J Rare Dis. (2018) 13:224
[7]Tian, et al. Human Genome Variation, 2016, 3: 15063.
[8]Xiaobei Guo, et al. Orphanet J Rare Dis. (2018) 13:224
[9]J Pediatr. Author manuscript; available in PMC 2010 January 25
[10]Am J Respir Crit Care Med Vol 186, Iss. 7, pp 593–597, Oct 1, 2012
[11]Farrell P M, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation[J]. The Journal of pediatrics, 2017, 181: S4-S15. e1.
[12]McCarthy C, et al. Personalised CFTR pharmacotherapeutic response testing and therapy of cystic fibrosis. Eur Respir J 2018; 51: 1702457
[13]Behan L, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J 2016; 47: 1103–1112.
[14]Genome Medicine. 2011;3(9):59.
[15]Leigh MW, et al. Genetics in medicine : official journal of the American College of Medical Genetics. 2009;11(7):473-487
[16]Eur Respir J 2017; 49: 1601090
[17]中华医学会儿科学分会呼吸学组疑难少见病协作组, 儿童原发性纤毛运动障碍诊断与治疗专家共识. 中华实用儿科临床杂志, 2018(2).
[18]Am J Respir Crit Care Med Vol 197, Iss 12, pp e24–e39, Jun 15, 2018
[19]Eur Respir J 2017; 49: 1601090
[20]Paediatric Respiratory Reviews 18 (2016) 8–17 http://dx.doi.org/10.1016/j.prrv.2015.07.017
[21]Guan Y, et al. Clinical and Genetic Spectrum of Children with Primary Ciliary Dyskinesia in China. Chest. 2020. https://doi.org/10.1016/j.chest.2020.06.045
[22]Chalmers JD, Alierti S and Blasi F. Management of bronchiectasis in adults. Eur Respir J 2015; 45: 1446–1462
[23]金贝贝等.原发性不动纤毛综合征四例并文献复习.中华结核和呼吸杂志, 2010, 33(3): 197-201
专家介绍

田欣伦
北京协和医院呼吸与危重症医学科副主任医师,硕士生导师;中国罕见病联盟呼吸病学分会副主任委员;中国研究型医院学会罕见病分会理事,美国NIH呼吸罕见病学者(Rare Lung Disease Scholar),中华医学会呼吸分会感染学组委员,中国医师协会呼吸医师分会疾病预防工作委员会委员,中国医师协会呼吸医师分会青年工作委员会委员;擅长呼吸系统常见病、支气管扩张、肺泡蛋白沉积症等疾病的诊治。对部分呼吸系统遗传病和罕见病有较深入研究。
本文由《呼吸界》编辑 大奔 整理,感谢田欣伦教授的审阅修改!
本文完
排版:Jerry