
 分享
分享

摘要
目的:研究2型及非2型支气管哮喘(简称哮喘)患者呼吸道共生微生物网络的作用特征。
方法:基于哮喘患者队列的前瞻性研究。采集2021年5月至2022年5月,首都医科大学附属北京朝阳医院呼吸与危重症学科收治的55例哮喘患者[男25例,女30例,中位年龄47.7岁(年龄范围34.3~63.0岁)]及来自首都医科大学附属北京朝阳医院体检中心的12名健康对照者的呼吸道诱导痰样本;根据呼出气一氧化氮(FeNO)水平,将哮喘患者分为高FeNO组22例(FeNO≥40 ppb,即2型哮喘组)、低FeNO组33例(FeNO<40 ppb,即非2型哮喘组)。对所有呼吸道诱导痰样本进行宏基因组二代测序并进行微生物群落多样性、组成特征、共生网络特征和代谢功能预测等生物信息学分析。采用Kruskal-Wallis秩和检验进行组间比较,采用线性判别分析(LEfSe)方法比较各组间的菌群组成差异。R语言用于微生物网络分析。此外,还采用PICRUSt预测微生物群落的代谢功能特征。
结果:健康对照组微生物群落中,厚壁菌门和变形菌门的占比(分别为29%和21%)均少于哮喘组患者(低FeNO组分别占37%和33%,高FeNO组分别占42%和26%)。低FeNO组的微生物网络有64对边,形成了16个群落;约75%的节点特征向量中心度的数值在0~0.05,25%的节点的特征向量中心度的数值在0.10~0.45;κ-核分解有4层,大约42%的顶点在中心的2层。高FeNO组的微生物网络有80对边,形成了18个群落;81% 的节点特征向量中心度的数值在0~0.05,19% 的节点的特征向量中心度的数值在0.10~0.35;κ-核分解有8层,21%的顶点位于中心的2层。低FeNO组和高FeNO组的主要功能差异表现在代谢途径(包括糖类、脂类、氨基酸和能量代谢)、抗药性、生物膜传输、信号传导、胞间通讯、细胞修复中。
结论:与非2型哮喘患者相比,2型哮喘患者的呼吸道微生物菌群的α-多样性更高,变形菌门的微生物含量更低,微生物网络更为聚集;两种内型哮喘患者的代谢功能预测结果有显著差异。
支气管哮喘(简称哮喘)是一种慢性气道疾病。根据是否存在2型炎症,哮喘可分为2型哮喘和非2型哮喘,2型哮喘以高呼出气一氧化氮(FeNO)、高嗜酸性粒细胞计数为特征 [ 1 , 2 ] 。这两种内型对糖皮质激素的治疗反应差别非常明显,其呼吸道微生态的特征可能是原因之一 [ 3 ] 。随着宏基因组二代测序以及生物信息学的发展,两种类型患者呼吸道的不同微生态特征及其与炎症类型的关联得以被揭示 [ 4 , 5 , 6 , 7 , 8 , 9 , 10 ] 。比如2型哮喘患者的呼吸道菌群中含有更高比例的放线菌门,且其含量与嗜酸性粒细胞计数明显正相关 [ 11 , 12 ] 。
除了菌群组成不同,呼吸道菌群之间彼此的相互作用也和哮喘的症状和炎症类型有关,其微生物共生网络的特征对于代谢和病理过程非常重要 [ 13 , 14 , 15 ] 。本研究的目的是探讨2型和非2型哮喘患者呼吸道共生微生物网络的特征差异。
对象与方法
一、研究对象
基于哮喘患者队列的前瞻性研究。本研究纳入2021年5月至2022年5月,首都医科大学附属北京朝阳医院呼吸与危重症学科收治的55例哮喘患者及北京朝阳医院体检中心体检的12名健康对照者,收集其呼吸道诱导痰样本。
哮喘患者纳入标准:(1)符合2024版《全球哮喘管理和预防策略》 [ 16 ] 中哮喘的诊断标准;(2)患者本人完全理解并同意本临床医疗研究的目的和实施,能配合医师完成诱导痰的采集。排除标准:(1)接受免疫抑制剂或长期类固醇激素治疗的恶性肿瘤、慢性自身免疫性疾病患者;(2)心肝脑肾等重要脏器严重受损者;(3)严重精神疾病患者;(4)妊娠或哺乳期患者。本研究已经过首都医科大学附属北京朝阳医院伦理委员审查会批准[(2020)425 号],参与本研究的哮喘患者及健康对照者均签署知情同意书。
根据FeNO水平,再将纳入的55例哮喘患者再分为高FeNO组22例(FeNO≥40 ppb [ 17 , 18 ] ,即2型哮喘组)和低FeNO组33例(FeNO<40 ppb,即非2型哮喘组)。
二、诱导痰采集和肺功能的测定
诱导痰采集步骤按照指南相关内容进行 [ 19 , 20 ] 。首先在雾化前10 min让患者吸入沙丁胺醇200~400 μg,以防止患者出现哮喘急性发作;嘱患者用清水漱口、擤鼻,以3%氯化钠注射液对患者雾化10~15 min后,嘱患者用清水漱口,将唾液尽量吐净,用力咳出深部的痰液到无菌容器中。如果此过程患者没有痰液咳出,则再次以4%氯化钠注射液雾化吸入5~10 min,再次嘱患者用力咳出深部的痰液到无菌容器中。痰液咳出后及时送检。
肺功能根据《中国国家肺功能测定指南》 [ 21 ] ,采用耶格Masterscreen PFT System肺功能仪进行测定。肺功能的测定时间为完成诱导痰操作前。
三、DNA 抽提和PCR扩增
根据E.Z.N.A. ® soil试剂盒(美国Omega Bio-tek公司)说明书进行总DNA抽提,DNA浓度和纯度利用NanoDrop2000进行检测,利用1%琼脂糖凝胶电泳检测DNA提取质量;用338F(5′ ACTCCTACGGGAGGCAGCAG 3′)和806R(5′ GGACTACHVGGGTWTCTAAT 3′)引物对V3~V4可变区进行PCR扩增,扩增程序为:95 ℃ 预变性3 min,27个循环(95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s),最后72 ℃延伸10 min(PCR仪:ABI GeneAmp ® 9700型)。扩增体系为20 μl,4 μl 5×tiFastPfu 缓冲液,2 μl 2.5 mmol/L dNTPs,0.8 μl 引物(5 μmol/L),0.4 μl FastPfu 聚合酶;10 ng DNA模板。
Illumina MiSeq测序:使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit(美国Axygen Biosciences公司)进行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测。利用QuantiFluor™-ST(美国Promega公司)进行检测定量。根据Illumina MiSeq平台(美国Illumina公司)标准操作规程将纯化后的扩增片段构建PE 2×300的文库。
构建文库步骤:(1)连接“Y”字形接头;(2)使用磁珠筛选去除接头自连片段;(3)利用PCR扩增进行文库模板的富集;(4)氢氧化钠变性,产生单链DNA片段。
利用Illumina公司的Miseq PE 300平台进行测序 [ 22 , 23 ] 。
四、微生物网络分析和统计学分析
采用SPSS 25.0软件,计量资料以 x¯±s 表示;计数资料以例(%)表示。组间比较采用Kruskal-Wallis 秩和检验,采用线性判别分析(LEfSe)方法来比较各组间的细菌组成差异,将 P值设为 0.05。用R语言 [ 24 , 25 ] 运行微生物组多变量关联线性模型(MaAsLins),以检验微生物组与临床变量之间的关联,包括以下R包:taxa [ 26 ] 、ggplot2、mix Omics [ 27 ] 、Tax4Fun2 [ 27 , 28 ] 、network [ 29 ] 。R的模块Igraph(version 1.2.5) [ 30 ] 用于微生物网络分析。此外,还利用PCRUST平台预测了京都基因和基因组百科全书(KEGG)中微生物群落的功能谱 [ 31 ] 。
1. 菌群共存网络的构建:首先,运用R语言里的corr.test函数分组计算了单个组别中所有菌种之间的Spearman相关性系数,得到相关性系数矩阵和 P值矩阵;并且采用Benjamini和Hochberg false discovery rate(FDR)方法矫正上述步骤中所得到的 P值。其次,基于Spearman相关性矩阵和校正后的 P值矩阵建立两组呼吸道菌群共存网络。Spearman相关系数和校正后的 P值的阈值分别为0.6和0.001。网络中的每一个节点(Node)代表一个菌种,连接节点之间每一个边(Edge)代表各菌种之间的相关性。菌群共存网络的构建是应用R语言中的graph_from_adjacency_ matrix函数。
2. 网络拓扑结构的基础分析:为了更好地了解所构建网络的拓扑结构,我们采用R语言中的igraph包中的相关函数计算并获得一系列基础的网络参数,其中包括边数(number of edges)、节点数(number of nodes)、连接性(connectance)、平均度(average degree)、平均介数(average betweenness)、平均路径长度(average path length)、平均最近邻度(average nearest-neighbor degree)、直径(diameter)、聚集系数(clustering coefficient)、介数中心性(betweenness centralization)、度中心性(degree centralization)、模块性(modularity)、模块数(number of modules)。此外,对网络中单个节点的度和介数中心性进行计算。应用网络k核分解方法计算并展示了两组菌群共存网络结构。
结果
一、受试哮喘患者特征描述
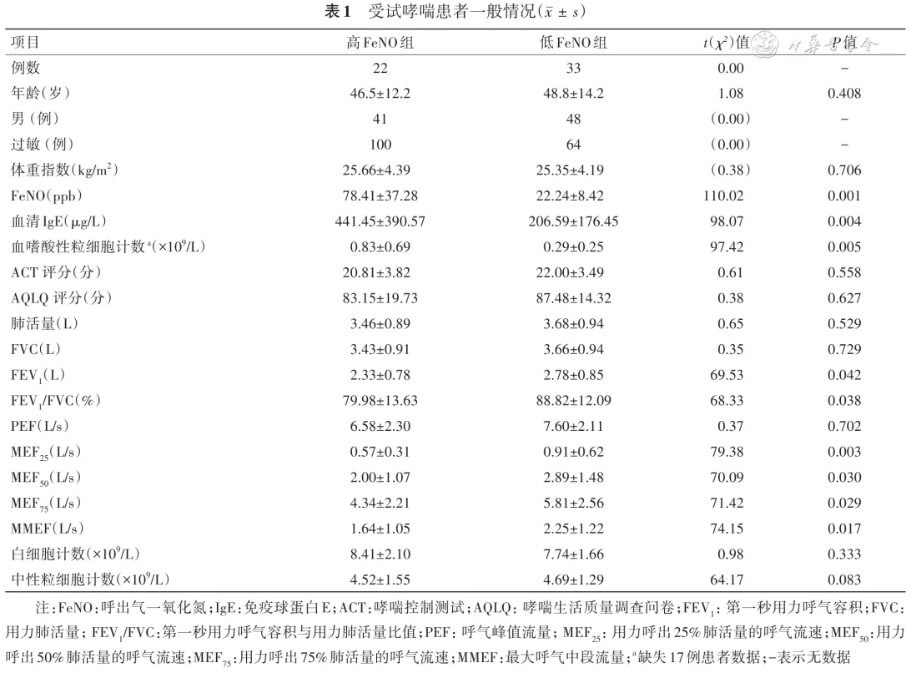
表1 列出了受试哮喘患者(高、低FeNO组)的临床指标。高FeNO组大多数小气道指数[用力呼出25%、50%、75%肺活量的呼气流速(MEF 25、MEF 50、MEF 75)及MEF 25/MEF 75]的数值均低于低FeNO组(均 P<0.05)。高FeNO组的血清IgE和血嗜酸性粒细胞计数显著高于低FeNO组(均 P<0.01)。高FeNO组和低FeNO的中性粒细胞计数、体重指数、ACT评分等方面差异无统计学意义。
二、菌群组成
不同组受试人群的诱导痰菌群组成如 图1 所示。与健康对照组相比,两组哮喘患者微生物群落中中观察到变形菌门的细菌丰度均更多。在门的水平,低FeNO组和高FeNO组的差异主要在厚壁菌门和变形菌门,包含了普雷沃菌属、韦荣球菌属、链球菌属、奈瑟菌属和嗜血杆菌属。在低FeNO 组,厚壁菌门和变形菌门分别占37%和33%;在高FeNO组,厚壁菌门和变形菌门则分别了42%和26%。而健康对照组厚壁菌门和变形菌门的占比均少于哮喘组患者(分别为29%和21%)。
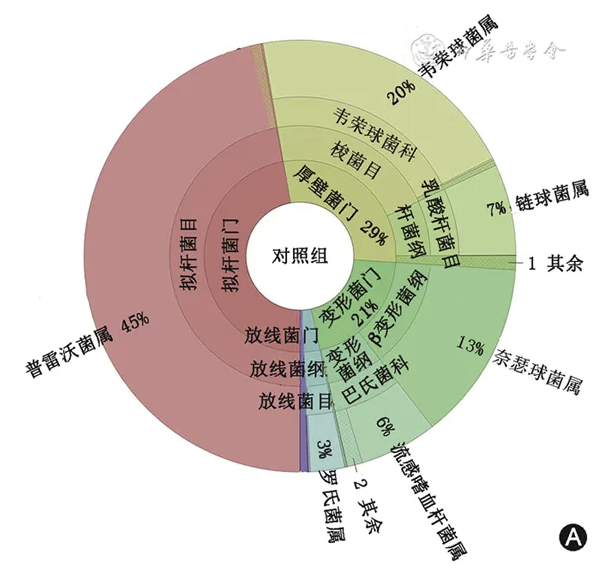
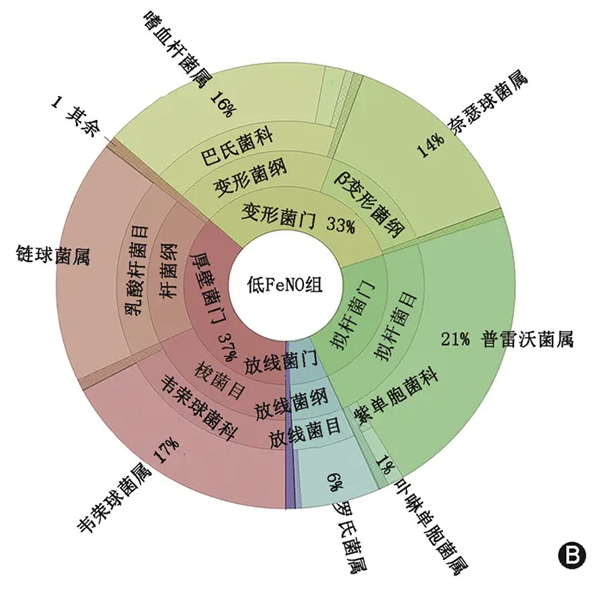
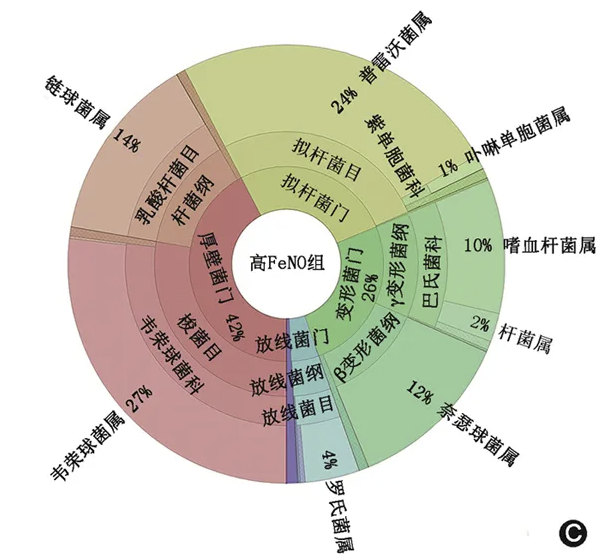
图1 不同组别诱导痰菌群组成 A.健康对照组;B. 低呼出气一氧化氮(FeNO)组;C. 高FeNO组
三、不同组别受试人群的微生物共生网络
我们以Spearman相关系数为基础,选择绝对值>0.6且显著性水平<0.001的关联构建了微生物共生网络。
如 表2 和 图2 所示,低FeNO组的微生物网络有64对边,形成了16个群落。高FeNO组的微生物网络有80对边,形成了18个群落。健康对照组的微生物网络有75对边,形成了12个群落。
表2 低、高呼出气一氧化氮(FeNO)组网络拓扑结构参数比较
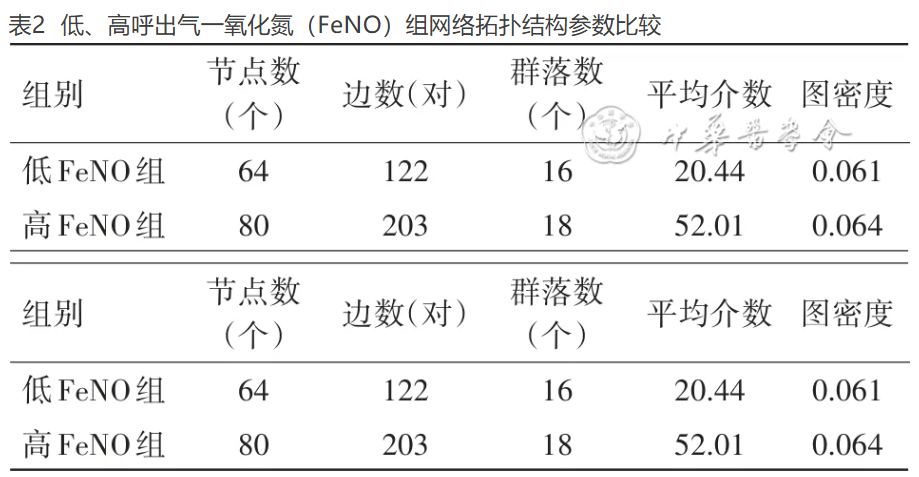
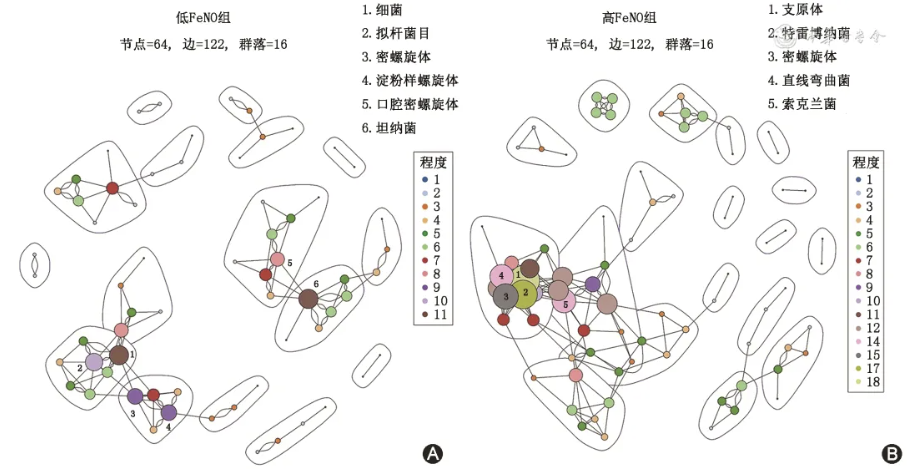
图2 低、高呼出气一氧化氮(FeNO)组微生物共生网络 A.低FeNO组;B.高FeNO组
如 表2 所示,低FeNO组和高FeNO组的微生物共生网络有较为接近的边数和节点数。但是,在一些网络参数对比上则差异明显。高FeNO组的微生物共生网络的所有参数都高于低FeNO组的微生物共生网络,表明高FeNO组的微生物共生网络更为密切和紧密。
如 图3 所示,低FeNO网络中节点的介数中心度明显低于高FeNO组网络,也就是说,低FeNO网络中的微生物彼此之间的重要性不如高FeNO组网络。此外,低FeNO网络中,约75% 的节点特征向量中心度的数值在0~0.05,25%的节点的特征向量中心度的数值在0.10~0.45。高FeNO网络中,81%的节点特征向量中心度的数值在0~0.05,19% 的节点的特征向量中心度的数值在0.10~0.35。即低FeNO组网络的微生物组分布更为中心,而高FeNO组网络中微生物群分布更为广泛。
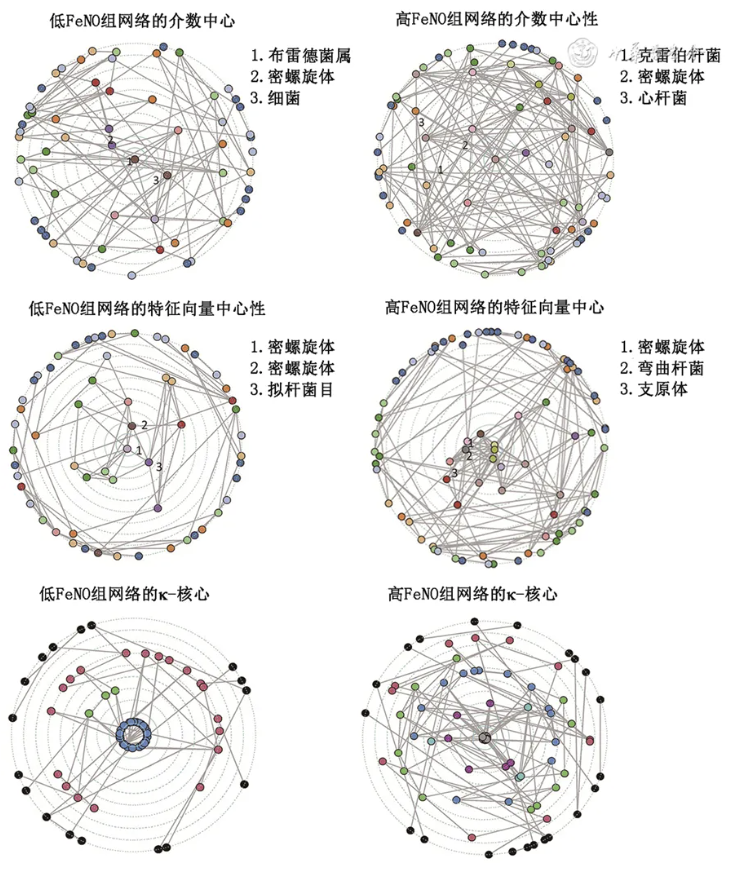
图3 低、高呼出气一氧化氮(FeNO)组的微生物共生网络参数化表达 A.介数中心性;B.特征向量中心度;C. κ-核分解结果
κ-核分解结果是根据其核心性在连续的层上显示顶点。越高的层表示核心度越高,在低FeNO组网络中有4层,并且大约42%的顶点在中心的2层中。在高FeNO组网络中,则有8层,21%的顶点位于中心的2层,即高FeNO组网络更为集中。
四、KEGG代谢功能预测
图4 显示了不同组别的功能预测结果,可见低FeNO组和高FeNO组的主要功能差异表现在代谢途径(包括糖类、脂类、氨基酸和能量代谢)、抗药性、生物膜传输、信号传导、胞间通讯、细胞修复中。
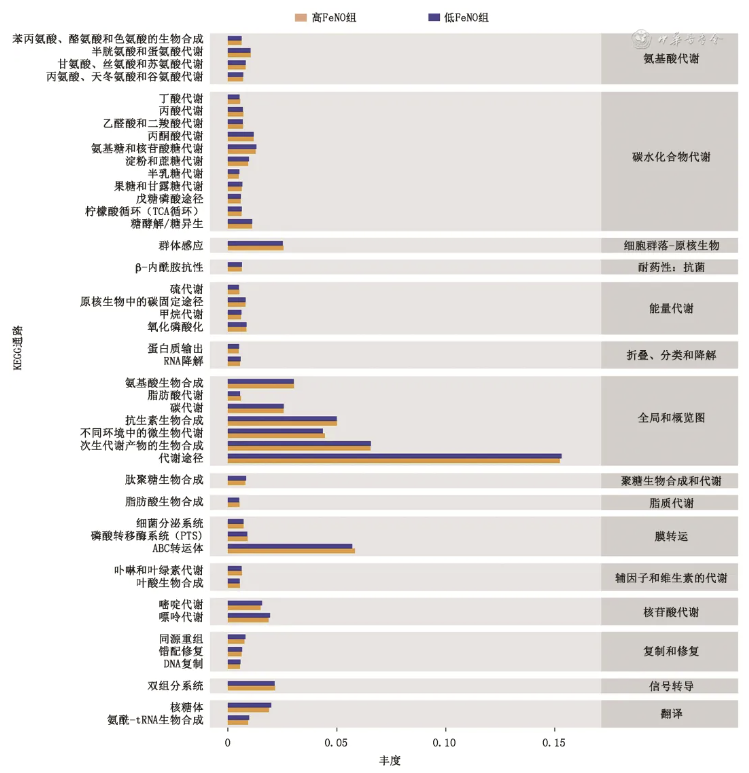
图4 高、低呼出气一氧化氮(FeNO)组功能预测的差异
讨论
本研究使用宏基因组二代测序方法鉴定微生物群,发现在健康对照者的下呼吸道中存在变形菌门、厚壁菌门、放线菌门、梭杆菌门和拟杆菌门等微生物群。哮喘是一种慢性气道疾病,表现出复杂的异质性,在环境因素和不同的易感基因相互作用下,具有很多不同的表型,而哮喘患者呼吸道微生物失衡就是影响表型的重要因素之一 [ 32 , 33 ] 。研究表明,哮喘患者的呼吸道微生物群和健康人相比存在显著不同。在控制良好的哮喘患者中,细菌多样性与支气管高反应性成反比。在生命早期,更高水平的变形菌门可能更容易诱导哮喘急性发作 [ 34 ] 。本研究和早期研究结果一致 [ 35 , 36 ] ,在门水平上,与健康对照组相比,高FeNO组和低FeNO组哮喘患者的变形菌门和厚壁菌门相关的细菌类群的丰度较高。
近年来,对于FeNO诊断哮喘价值的临床研究显示,如果FeNO水平为9~15 ppb,其诊断哮喘的敏感度可以达到85%~96%,但是特异度仅为13%~48%。如果FeNO水平提高到47~76 ppb,则其诊断哮喘的特异度可以提高到88%~100%,但敏感度则显著降低至13%~56%。可以看出,FeNO的水平越高,诊断准确性越高 [ 37 , 38 ] 。如果FeNO的水平≥40 ppb,诊断2型哮喘的准确性超过90%。因此我们以FeNO水平40 ppb为分组标准来区分2型哮喘患者和非2型哮喘患者。本研究结果发现不同类型哮喘患者不仅在呼吸道微生物组成有显著差异,其微生物的共生网络参数及预测功能也有很大区别。与高FeNO组比较,低FeNO组的变形菌门含量显著高,包括嗜血杆菌属和奈瑟菌属,厚壁菌门则显著低,这与国内外其他研究一致 [ 39 , 40 , 41 ] 。
既往关于微生物网络的研究主要集中在肠道共生菌群,而关于呼吸道微生物的网络研究则罕见报道。肠道微生物群落的研究表明,微生物之间的关联在决定群落的整体结构和功能方面发挥着重要作用,对微生物网络的各种拓扑特性的分析可以使我们探讨微生态的有关特征。从门的水平上说,肠道菌群中拟杆菌门、厚壁菌门、变形菌门、放线菌门、疣菌门是5个最具优势的菌门,其中拟杆菌门和厚壁菌占总相对丰度的80%以上 [ 42 ] 。拟杆菌是健康人类肠道微生物组内生态系统的重要驱动因素。胃肠道微生物与人类炎症性肠病、肠易激综合征、肥胖、糖尿病、肝炎和孤独症等疾病相关。与患病人群相比,健康人群中拟杆菌门-拟杆菌门和厚壁菌门-厚壁菌门的相互作用更丰富,这表明同一菌群之间的相互作用在维持肠道稳态方面发挥重要作用。在相关的疾病网络中,相互作用最丰富的门是变形杆菌门,并且常见变形菌门-放线菌门的相互作用,这可能是肠道微生态失调的标志 [ 43 ] 。除此之外,肠道微生物网络还与毒素产生、蛋白质分泌、厌氧代谢、核酸代谢、细胞分裂、信号转导等相关。本研究表明呼吸道微生物的网络相互作用很可能也与呼吸道疾病相关。
除2型炎症外,由Th17细胞驱动的独有炎症类型可能形成非2型哮喘内型的基础。Th17细胞的特征在于分泌IL-17A、IL-17F、IL-21和IL-22,Th17细胞通过IL-17A和IL-17F促进中性粒细胞募集,并作用于哮喘中的非2型途径。Huang等 [ 7 ] 鉴定了一组主要与Th17炎症相关基因的上皮表达显著相关的变形菌,证明了变形菌门的丰度与Th17/IL-17介导的炎症相关。IL-17水平升高与皮质类固醇抵抗、气道高反应性、黏液分泌过多、气道阻塞有关。本研究中,低FeNO组较高FeNO组的变形菌门菌群丰度更高,和既往研究的结果较为一致。
存在于呼吸道内的微生物群落与代谢功能相关。与低FeNO组相比,高FeNO组的微生物网络更为密切和丰富,有关核酸代谢、脂类代谢、能量代谢、细胞修复及抗药性的功能也显著更强。本研究的结果可为临床诊疗提供有益的基础信息 [ 44 , 45 , 46 ] 。
本研究纳入哮喘患者例数较少,今后需增加入选研究样本量,纳入更多的临床数据进行前瞻性研究。研究应进一步减少导致呼吸道生态失调的影响因素,如各种哮喘治疗药物(包括吸入和口服糖皮质激素),环境因素(如室内和室外污染)和饮食等。气道微生物组成和共生网络的差异可能影响不同的哮喘炎症内型。我们的研究结果表明,今后哮喘可以采用更多具有针对性的治疗方法。
作者:王雯 王斐然 郭越 张惠冰 蒋芳帆;通信作者:王雯,首都医科大学附属北京朝阳医院呼吸与危重症学科
引用本文: 王雯, 王斐然, 郭越, 等. 2型及非2型支气管哮喘患者呼吸道共生微生物网络的作用特征[J]. 中华结核和呼吸杂志, 2024, 47(12): 1121-1129. DOI: 10.3760/cma.j.cn112147-20241015-00611.
本文转载自订阅号「中华结核和呼吸杂志」
原链接戳:【论著】2型及非2型支气管哮喘患者呼吸道共生微生物网络的作用特征
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry