
 分享
分享

近日,北京大学人民医院陈宏斌副研究员(第一作者)、王辉教授(通讯作者)等在npj Biofilms and Microbiomes发表了题为《Integrating respiratory microbiome and host immune response through machine learning for respiratory tract infection diagnosis》的最新研究成果。

下呼吸道感染(Lower respiratory tract infections,LRTIs)是全球导致人口死亡的主要原因之一,每年夺走超过300万人的生命。然而,传统的微生物学检测在急性呼吸道的诊断上依旧存在诸多不足,如敏感性低、检测速度慢以及检测范围有限。这些问题导致我们在处理大量病例时难以准确识别病原体。这不仅导致了不必要的抗生素使用,还加剧了抗生素耐药性增加的问题。
为了寻找更有效的诊断方法,本研究收集了2020年至2021年间136例下呼吸道感染患者的支气管肺泡灌洗液(BALF)标本,并运用元转录组学技术分析了下呼吸道微生物组(LRTM)和宿主反应。研究结果显示,LRTIs患者的LRTM多样性显著降低,表现为正常微生物群落的减少和条件致病菌的增加。在LRTIs组中,上调的的差异表达基因(DEGs)主要富集于感染免疫反应相关的通路。肺炎克雷伯菌在LRTIs中的丰度显著增加,并与宿主感染或炎症基因TNFRSF1B,CSF3R和IL6R紧密相关。基于LRTM和宿主转录组数据,本研究构建了一个机器学习模型,该模型通过12个筛选特征有效区分LRTIs和非LRTIs。验证集数据显示,随机森林训练的模型具有最佳性能(ROC AUC:0.937,95% CI:0.832-1)。独立外部数据集显示该模型的准确率为76.5%。本研究表明,整合LRTM和宿主转录组数据的模型有望成为诊断LRTIs的一种有效工具。
1、受试者基线特征
本研究回顾性收集了68例在北京大学人民医院就诊的疑似LRTIs患者临床和实验室信息用于机器学习研究。在这些患者中,大约41例被确诊为LRTIs,包括训练队列中的28例和验证队列中的13例。其余的27例患者被确诊为非LRTIs,包括训练队列中的18例和验证队列中的9例。随后,为了对预测模型进行外部验证,我们另外招募了68例疑似LRTIs患者,其中45例被确诊为LRTIs,23例被确诊为非LRTIs。利用SPSS 26软件,我们对LRTIs组和非LRTIs组的基线人口统计信息、临床参数和实验室特征进行比较,并计算了相应的P值。结果发现LRTIs患者和非LRTIs患者的关键临床特征差异不大(P>0.05)。
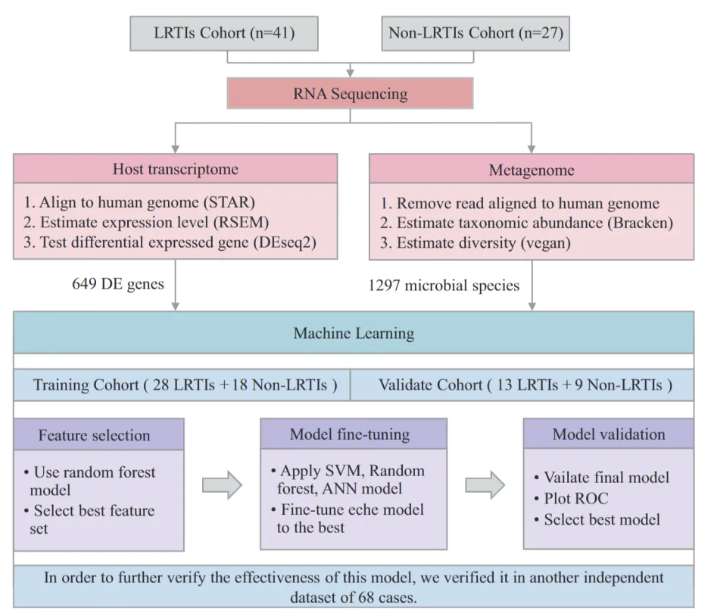
图1.研究流程图
2、LRITs患者与非LRITs患者的下呼吸道微生物组的差异
本研究发现,与非LRTIs患者相比,LRTIs患者BALF中的微生物群落多样性显著降低。此外,在非LRTIs组中,正常菌群如牙龈卟啉单胞菌、牙棒杆菌和密螺旋体的丰度显著增加,而在LRTIs组中,条件致病菌如肺炎克雷伯菌、铜绿假单胞菌、肺炎链球菌、耶氏肺孢子菌等的丰度显著增加。这些结果表明,LRTM多样性的降低和条件致病菌丰度的增加是LRTIs发生的的高风险因素。
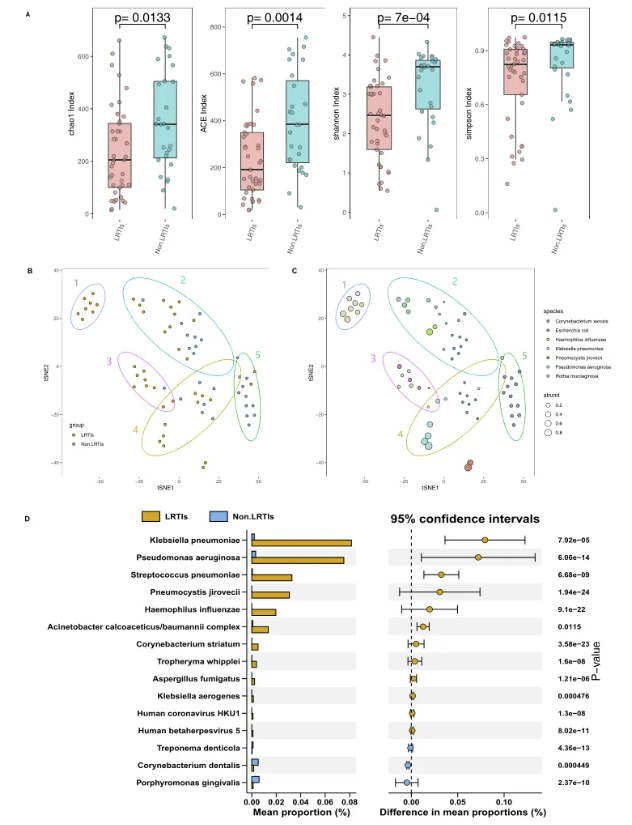
图2. LRITs患者与非LRITs患者的下呼吸道微生物组的差异
3.LRTIs患者与非LRTIs患者宿主转录组差异
通过对BALF宿主转录组进行分析,本研究发现LRTIs组共有649个差异表达基因,其中613个基因表达上调,36个基因表达下调。LRTIs组中上调的DEGs数量显著多于非LRTIs组。这些宿主DEGs被划分为三个不同的簇,主要在免疫系统、信号传递、传染病(包括病毒和细菌)、信号分子和相互作用以及其他通路上富集。需要注意的是,簇C的信号分子和相互作用通路中富集的DEGs数量显着高于簇A和簇B中的DEGs。
在基因本体(GO)富集分析中,DEGs主要在感染免疫相关通路中富集,特别是LRTIs组中上调的DEGs主要富集在白细胞相关通路中。在京都基因与基因组百科全书(KEGG)富集分析中,LRTIs组中的DEGs主要富集在细胞因子-细胞因子受体相互作用、NF-κB信号通路、中性粒细胞胞外陷阱形成、趋化因子信号通路、TNF信号通路、HIF-1信号通路以及Th17细胞分化。利用clusterProfiler工具,我们对DOSE数据库中的DEGs进行了疾病富集分析。结果显示,这些DEGs主要与肺部感染相关疾病,如肺炎等,有着显著的富集关系。
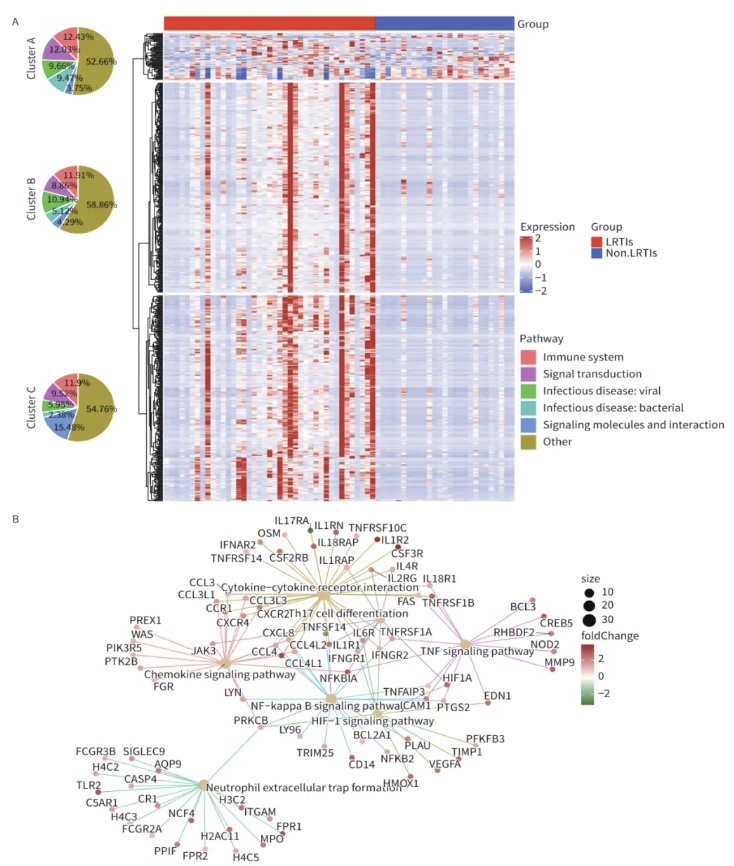
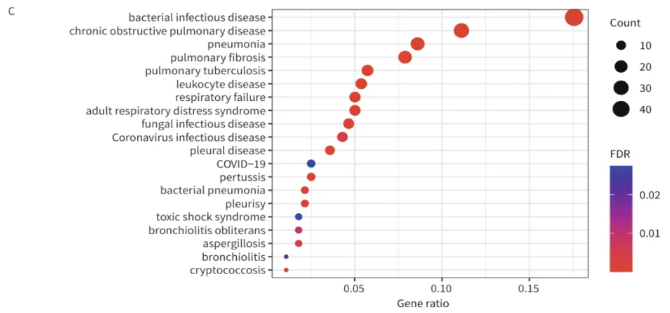
图3. LRTI患者与非LRTI患者的宿主转录组差异
4.下呼吸道微生物组与宿主基因之间的相互作用
在LRTIs中,宿主基因与LRTM之间的相互作用发挥着重要的作用。为此,我们对宿主差异表达基因与LRTM之间的相关性进行了深入分析。研究发现,有42个宿主基因与条件致病菌呈正相关,而与正常菌群呈负相关。另外,4个宿主基因与条件致病菌呈负相关,而与正常菌群呈正相关。进一步地,本研究对特定微生物基因和宿主差异表达基因进行了相关性分析,发现了肺炎克雷伯菌的α2-巨球蛋白家族蛋白与宿主基因TNFRSF1B、CSF3R和IL6R之间的强相关性。这些发现为理解LRTIs中宿主与微生物之间的复杂互动提供了新的视角,并可能为未来的诊断和治疗策略提供潜在的靶点。
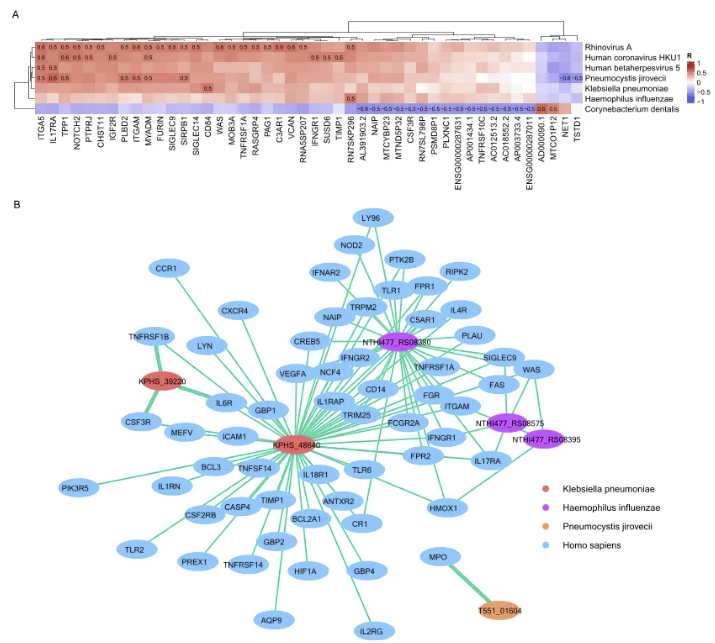
图4.活跃下呼吸道微生物组与宿主基因之间的相互作用
5.整合下呼吸道微生物组与宿主应答区分LRTIs和非LRTIs
正如前文所述,LRTIs并非由单一微生物引起,而是LRTM与宿主相互作用的结果。在传统的临床实践中,LRTIs的诊断往往依赖于患者的临床表现、影像学检查和炎症标志物。本研究将呼吸道微生物群和宿主的免疫反应纳入诊断模型,有望为临床医生提供更准确的LRTIs和非LRTIs区分手段。
本研究利用随机森林算法筛选出12个关键特征(包括6个肺部差异微生物群落特征和6个宿主差异表达基因特征 )以区分LRTIs和非LRTIs。研究结果表明,整合了LRTM和宿主基因特征的模型展现出卓越的性能,其受试者工作特征曲线下面积(ROC AUC)达到了0.937(95%置信区间:0.832-1)。在独立的外部数据集上,该模型的准确率达到了76.5%。这些发现表明,结合微生物组和宿主转录组数据的模型在临床上具有潜在的应用价值,有助于提高LRTIs的诊断准确性和效率。
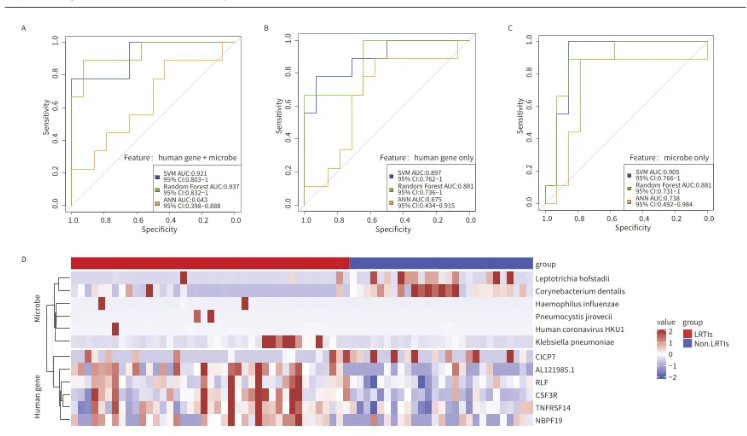
图5.根据下呼吸道微生物组与宿主反应区分LRTIs和非LRTIs
本研究表明,通过整合下呼吸道微生物组(LRTM) 和宿主转录组数据构建的模型,有望成为诊断LRTIs的一种有效工具。这种模型的应用有助于临床医生更精确地诊断LRTIs,进而为患者提供更为针对性的治疗方案,从而减少抗生素耐药性的发生风险,对改善公共卫生和促进抗生素的合理使用具有重要意义。
该研究得到了国家重点研发计划(2022YFA1304300)和北京市科委计划(Z191100006619100)的支持。
作者简介

第一作者:陈宏斌
理学博士,硕士研究生导师,2017-2018年University College London访问学者研修生物信息学。现任北京大学人民医院检验科副主任,中华医学会检验分会青年学组委员,中华医学会微生物学与免疫学分会青年委员会委员,中华医学会检验分会临床微生物学组委员等。以第一作者或通讯作者在Genome Medicine、Cell reports、Clinical Infectious Diseases、Journal of Advanced Research、Clinical Chemistry、Science Bulletin等发表SCI文章28篇,总IF162分。

通讯作者:王辉
医学博士,博士生导师,国家杰出青年基金获得者、国家卫健委突出贡献中青年专家。现任北京大学人民医院检验科主任,北大医学部检验学系主任,中国医促会临床微生物分会主任委员、中华医学会检验分会常委兼临床免疫学组长、中华医学会微生物学和免疫学分会常委兼临床微生物学组组长等。研究方向:细菌耐药、临床检验诊断学。
作者:陈宏斌、王辉、吴万云(北京大学人民医院);审校:陈宏斌(北京大学人民医院)
本文转载自订阅号「京港感染论坛」
原链接戳:【前沿速递】北京大学人民医院王辉教授团队:通过机器学习整合呼吸道微生物组和宿主免疫应答以诊断呼吸道感染
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry