
 分享
分享

摘要
伯特-霍格-杜布综合征(Birt-Hogg-Dubé syndrome,BHD)是一种罕见的常染色体显性遗传性疾病,其临床特征为弥漫性肺部囊状病变、自发性气胸、皮肤纤维毛囊瘤或毛盘瘤和多种类型的肾脏肿瘤。BHD综合征发病机制与第17号染色体的卵泡素蛋白(folliculin,FLCN)相关基因功能丢失变异有关。近年来我国BHD综合征病例报道数量逐年攀升,与欧美高加索人群比较,BHD综合征中国人群多累及肺部,主要呈弥漫性囊状病变并常导致气胸,而肾脏和皮肤受累相对少见,故临床上漏诊和误诊较普遍,缺乏规范诊疗与科学管理。为进一步提高BHD综合征诊治和管理水平,特邀请全国相关领域专家制定本专家共识。本共识通过投票形成15条专家推荐意见,具体包括临床评估、诊断标准、鉴别诊断、治疗、随访以及家系管理等,特别是根据我国实际国情,优化了诊断标准。旨在通过本专家共识,推动BHD综合征规范诊疗和科学研究,改善患者预后。
一、概述
伯特-霍格-杜布综合征(Birt-Hogg-Dubé syndrome,BHD)又称Hornstein-Knickenberg综合征,是一种罕见的常染色体显性遗传性疾病,其临床特征为肺部弥漫性囊状病变、或伴自发性气胸、皮肤纤维毛囊瘤或毛盘瘤和多种类型的肾脏肿瘤[1]。BHD综合征的发病与第17号染色体的卵泡素蛋白(folliculin,FLCN)相关基因功能丢失变异有关[2, 3]。
近年来,随着我国临床医师对BHD综合征认识的不断提高,病例不断见诸报道[4, 5, 6, 7, 8, 9, 10, 11]。与欧美高加索人群比较,亚洲人群BHD综合征多累及肺部,表现为双侧、多发肺部囊状病变,有自发性、复发性气胸风险,而肾脏和皮肤受累报道相对较少,因此临床上漏诊和误诊并不少见[3, 4,12, 13],缺乏正确规范的诊疗和科学管理。为进一步提高BHD综合征诊治和管理水平,来自全国各地、多学科组成的BHD综合征诊治和管理专家共识编写专家组,检索国内外相关文献并在充分复习重点文献、评估国内外相关研究进展的基础上,结合我国实际情况,讨论制定本专家共识(以下简称“共识”)。旨在通过本共识,推动该病规范诊疗、全程管理及科学研究,改善患者预后。
共识在国际实践指南注册与透明化平台注册(http://www.guidelines-regestry.org,注册编号:PREPARE-2023CN283)。专家组收集临床医务工作者在BHD综合征诊疗过程中最为关注的临床问题进行了多轮讨论,在方法学专家指导下评估国内外证据,并结合我国国情,采用Delphi法经过专家投票(附录表1)最终确定15条推荐意见,内容包括临床评估、诊断、鉴别诊断、治疗、随访及家系管理等,特别是根据我国国情,改良并优化了诊断标准。对于存在分歧的推荐意见,至少需要≥50%参与者认可且持相反意见的比例<20%,未满足此标准则不形成推荐意见。
二、流行病学和疾病负担
BHD综合征的全球确切发病率与患病率尚不清楚,根据BHD综合征基金会相关数据,截至2017年,全球已报道超过600个BHD综合征家系。采用不同统计计算方法,BHD综合征的发病概率差异较大。Muller等[14]通过自发性气胸流行病学数据和贝叶斯定理计算出普通人群中BHD综合征的患病率约为1.86/1 000 000。最近美国Savatt等[15]对13.6万人进行全外显子测序,发现35例患者带有FLCN基因的致病和(或)可能致病变异(1/3 885),其中23例患者具有BHD综合征相关临床表现,但只有4例(4/35,11.4%)既往诊断为BHD综合征,提示其发病率被严重低估。截至2021年底,我国已报道来自143个家庭的287例患者[12]。
三、临床表现
1. 肺部表现:肺是BHD综合征最常累及的脏器之一,亚洲国家BHD综合征肺部受累发生率高于欧美国家[4,13]。以肺部受累为首发表现患者超过60%[9]。肺部受累主要表现为弥漫性囊状改变和自发性气胸。85%~98%的亚洲BHD综合征患者存在肺部囊状改变[4,9,12, 13,16, 17],大多数患者没有症状,但发生自发性气胸的风险较高,58%~71%患者有自发性气胸病史,首次气胸多发生在40岁前[13,18]。少量气胸可以无症状,严重者可出现呼吸困难和胸痛、呼吸音减弱甚至消失。BHD综合征患者气胸发生风险是无BHD综合征家族成员的50倍[19]。近年来国内外学者进一步分析了FLCN基因变异与气胸发生的相关性。一项德国队列研究显示FLCN致病变异基因c.250-2A>G(77%)和c.1300G>C(59%)较变异基因c.1285dup(37%)显著增加了BHD综合征发生气胸的风险[20]。最新Wang等[21]分析安徽省76例BHD综合征基因和表型的相关性发现,1~3号外显子大片段缺失和点变异的气胸发生率分别为91%和 58%,P值0.047,差异存在统计学意义。BHD综合征对肺功能的影响较小,引起低氧或呼吸衰竭并不常见[22]。
2. 皮肤表现:皮肤病变主要表现为纤维毛囊瘤、毛盘瘤,可伴有软垂疣。纤维毛囊瘤和毛盘瘤是单一病理过程的不同阶段。纤维毛囊瘤是该病的特征性表现,出现在欧美90%的皮肤损害的BHD综合征患者中,常分布在面部、颈部、躯干上部[23],见图1。纤维毛囊瘤为多发、肤色或略白色、圆顶状、直径为1~5 mm的丘疹,其数量通常在5~100个不等。组织学上,表皮大致正常,真皮可见呈囊状扩张的毛囊样结构,从中央毛囊发出多个吻合的上皮细胞条索,形成类似脚手架外观[24],被成纤维细胞围绕。82%~92%的欧美患者存在皮肤病变,而我国患者皮肤病变发生率约18%~68%[9,13]。皮肤病变多在成年后发现,伴随年龄增长发病率增加,且有数量增多趋势。日本的研究结果显示,患者年龄>30、50和60岁的皮肤病变发生率分别为50%、70%和100%[25]。
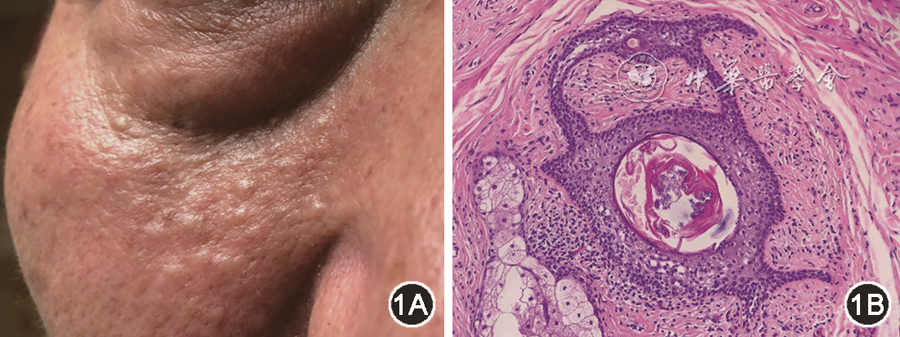
图1 BHD综合征患者的面部纤维毛囊瘤,图1A可见鼻旁区域多发、肤色的小丘疹;图1B为纤维毛囊瘤组织病理切片图像(HE中倍放大),可见增殖的毛囊上皮及周围富含纤维细胞的间质组织
3. 肾脏表现:约25%~35%的BHD综合征患者会发生肾脏肿瘤,通常为双侧、多灶性且生长缓慢;肿瘤诊断的中位年龄为46~50岁[19,23,26, 27]。最常见的肿瘤病理类型是混合性嗜酸-嫌色细胞肿瘤(50%)、嫌色细胞癌(34%)和嗜酸细胞瘤(9%),见图2。少见类型包括透明细胞癌和乳头状肾癌。此外,患者肾脏的另一个特点是“嗜酸细胞增多”,可表现为镜下肾实质中散在分布的微小嗜酸细胞聚集病灶,可能是肾肿瘤的癌前病变[28]。我国报道的肾脏受累发生率较低,其中肾癌约7.2%,肾脏血管平滑肌脂肪瘤约1.4%,肾囊肿约8.6%[12]。与von Hippel-Lindau 综合征(VHL综合征)等其他遗传性肾癌相比,BHD综合征的肾肿瘤出现远处转移的概率较低,可能与该病嫌色细胞癌为主的病理类型相关。
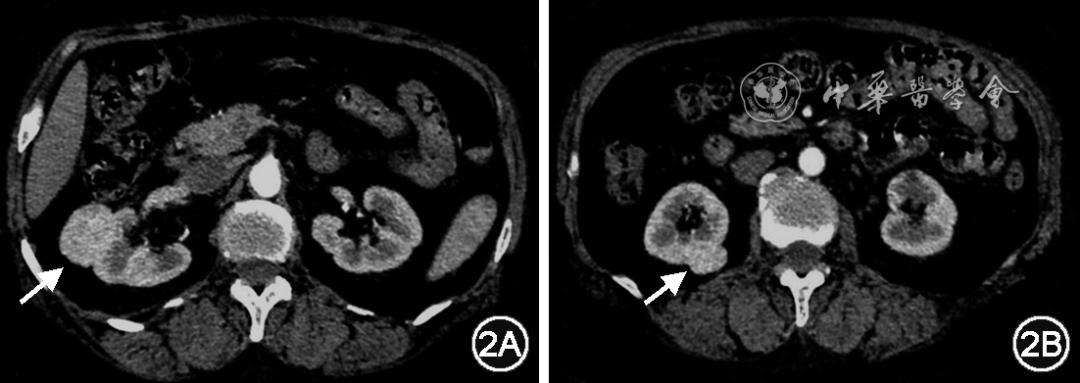
图2 64岁女性BHD综合征患者的肾脏肿瘤,肾脏增强CT轴位图示右肾见两枚类圆形软组织肿块(图2A和图2B箭头所示),边界清晰,密度均匀,均匀强化;手术病理证实均为嫌色细胞癌
4. BHD综合征少见的临床表现:主要包括结肠癌[29]、支气管肺癌[30]等恶性肿瘤。这些表现是否真正与BHD综合征相关仍有待确定[2,31]。
【推荐意见1】FLCN是目前唯一确定的BHD综合征致病基因,对家系样本进行测序分析有助于快速鉴定致病基因变异。可根据患者实际情况选择一代测序、MLPA或NGS等变异检测方法。(投票专家43人;赞成43票,反对0票,弃权0票)。
四、FLCN基因及分子诊断方法
FLCN基因位于染色体17p11.2上,包含14个外显子,翻译起始密码子位于外显子4。FLCN蛋白由579个氨基酸组成,在包括皮肤、肺脏、肾脏等大多数组织中表达[32]。FLCN蛋白是一个保守但不含已知结构域的蛋白,其功能尚待阐明。迄今为止,对FLCN功能研究支持其在多种代谢途径和细胞过程中的作用,包括mTORC1途径的调节、PGC1α和线粒体生物发生的调节、细胞间黏附和RhoA信号传导等(图3)。
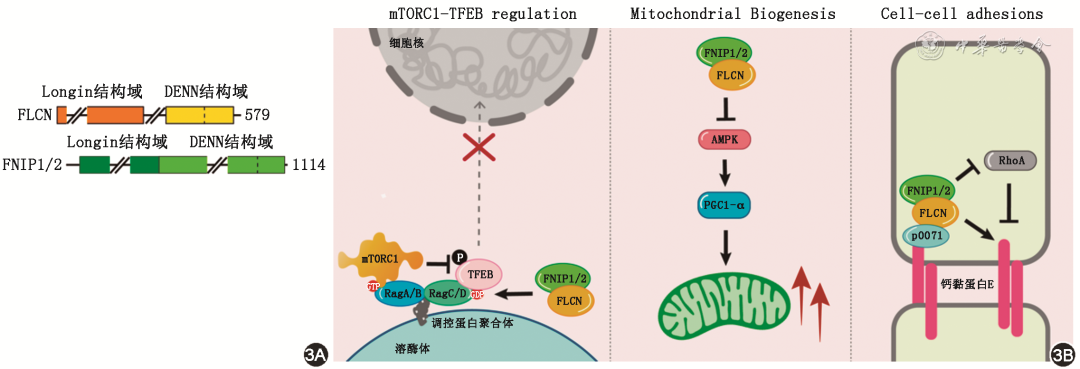
图3 BHD综合征的发病分子机制,图3A为卵泡素蛋白(FLCN)与其伴侣蛋白(FNIP1/2)的结构域组织;图3B为FLCN蛋白所涉及的生物学过程的简略示意图:(左)作为RagC/D的GAP,促使mTORC1磷酸化转录因子EB(TFEB)进而负向调控TFEB的入核转录过程;(中)抑制能量应激激酶AMPK的活性,负向调控PGC1-介导的线粒体生物发生过程;(右)抑制细胞骨架肌动蛋白调节分子RhoA活性,同时促进上皮钙黏蛋白E(E-cadherin)活力,进而增加细胞黏附能力
FLCN是目前唯一确定的BHD综合征致病基因,在患者检测出该基因的致病胚系杂合变异是BHD综合征的确诊金标准[33]。研究表明携带胚系杂合变异同时伴随体细胞变异或杂合性丢失的患者更易出现肾癌等肿瘤的表型[34]。
有研究认为,FLCN失活变异导致TFE3/TFEB过度激活可能是引发BHD综合征的主要原因[35, 36]。在小鼠肾脏特异性敲除FLCN基因促发肾脏囊性或实体瘤,可复制出BHD综合征患者肾癌表型;同时在小鼠肾脏敲除FLCN和TFEB基因能够阻遏肿瘤发生。
已经鉴定的FLCN变异可见于整个基因包括编码区和非编码区。根据人类基因突变数据库(professional 2022.4)记录,FLCN致病变异共265种,按照频率高低分布如下:小片段缺失(32.8%),无义变异(17.4%),剪接变异(14.0%),大片段缺失(11.3%),错义变异(10.2%),小片段插入(9.8%),小片段缺失及插入(3.4%),大片段插入(0.8%)和调控区域变异(0.4%)。其中热点变异为11号外显子polyC缺失或重复(285delC或c.1c.1285dupC),中国汉族人群BHD综合征患者携带c.1285delC或c.1285dupC变异可高达1/4[12]。FLCN致病性变异的判定依据包括家系分析和分子检测结果。移码和无义变异、基因内大片段缺失或重复等可导致FLCN蛋白失活的变异可判定为致病变异,而非经典剪接位点变异和错义变异需通过进一步的实验和多个家系分析确定变异的致病性[37]。FLCN基因一代测序(sanger sequencing)和多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)以及二代测序(next-generation sequencing,NGS)包括靶向基因包(Targeted gene panel)、全外显子组测序(whole exome sequencing,WES)和全基因组测序(whole genome sequencing,WGS)方法均可用于BHD综合征致病基因变异鉴定。由于NGS检测通量非常高,还能鉴定包括大片段缺失或重复、影响mRNA剪接的深度内含子变异等复杂变异,且近来检测成本降低,已成为临床基因变异检测的主流技术[38]。
五、影像学
1. 常用影像学检查方法。(1)X线胸片:主要用于观察气胸;(2)胸部CT:主要用于观察肺部囊状病灶及气胸;(3)腹部超声、CT/MR平扫及增强扫描:主要用于观察肾脏病变。由于超声对肾脏肿瘤的准确度较低,CT/MR平扫及增强扫描是诊断肾脏肿瘤的主要影像学检查方法,尤其是增强扫描对肿瘤类型的诊断及鉴别诊断至关重要。而反复CT检查存在X线辐射风险,所以MR检查是BHD综合征患者监测肾脏病变推荐使用的影像学检查手段。
【推荐意见2】BHD综合征患者应行胸部CT评估肺部囊状病变,常规评估肾脏有无肿瘤病灶,可以酌情选择下列检查方法(超声、增强CT或MR)。(投票专家43人;赞成43票,反对0票,弃权0票)。
2. BHD综合征临床关键问题的影像学评价。(1)肺部病变:①主要表现为囊状改变,常为双侧、多发、大小不一(病灶直径从几毫米到数厘米或更大)。肺部囊状病变通常表现为薄壁,形状不规则(以圆形、椭圆形和扁平状多见),部分可融合,常分布于双肺基底部、中下肺、邻近叶间裂和纵隔,多位于两肺邻近纵隔胸膜下[39, 40],见图4;CT多平面重建图像有助于观察全肺囊状病变的分布特点;②合并气胸:气胸常为BHD综合征患者就诊的首发症状[12]。BHD综合征患者囊状病变的最大直径、最大体积与发生气胸的风险密切相关[40],CT表现为胸膜腔内出现气体影,见图4。(2)肾脏病变:BHD综合征患者肾脏肿瘤发病率可能与种族及基因变异类型有关,主要表现为双侧、多发的混合性嗜酸细胞瘤(嗜酸细胞瘤和嫌色细胞混合类型)[28,41],有报道可发生肾脏血管平滑肌脂肪瘤[9,42],也有少数合并透明细胞癌的报道[12]。(3)影像报告中应包括的内容:①胸部CT报告中应重点描述BHD综合征患者肺部是否存在囊状病变及其分布的部位、形态、大小等特征,同时注意观察是否存在气胸;②腹部超声、CT及MR应重点关注患者肾脏是否存在病变,对于可疑肾脏肿瘤患者应行肾脏动态增强CT或MR扫描,描述其部位、大小、强化特征及与周围组织的关系。
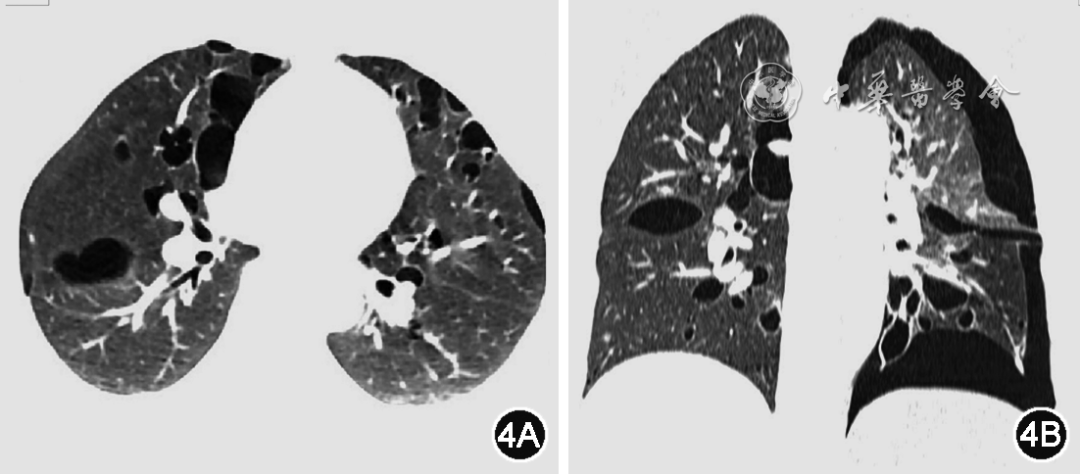
图4 43岁女性BHD综合征患者的肺部囊状病灶,图4A为常规CT横断位图像,显示两肺多发薄壁椭圆形或不规则形囊状病灶,边界清晰;图4B为患者1年后并发左侧气胸时CT冠状位重建图像,显示两肺多发的含气囊状病灶,邻近纵隔胸膜下及斜裂区分布为著,左侧胸腔内见气体影
【推荐意见3】临床医师应根据相应的临床表现,并结合基因检测结果或者BHD综合征家族史建立诊断。(投票专家43人;赞成43票,反对0票,弃权0票)。
六、诊断标准
BHD综合征由于其罕见性,常被误诊为自发性气胸、肺大疱、淋巴管肌瘤病(lymphangioleiomyomatosis,LAM)、肺朗格汉斯细胞组织细胞增生症(pulmonary langerhans cell histiocytosis,PLCH),或其他弥漫性囊状肺部疾病(diffuse cystic lung disease,DCLD)[43]。此外,其相对较轻但多样的临床表现,使其较难早期筛查、早期识别、早期诊断,故临床上常存在误诊与漏诊。在缺乏相关诊疗经验的医疗单位,一旦临床疑诊,建议转诊至具有相应诊治经验和条件的医疗机构进一步诊治。2009年欧洲BHD综合征联盟提出的诊断标准,临床应用广泛,但我国患者皮疹识别率低,且常常难以进行活检证实。因此,本共识专家组结合我国BHD综合征的临床特点和实际国情,对该诊断标准进行了优化,增加一级亲属家族史作为遗传学标准,调整皮疹为临床标准第二条,详见表1[33]和图5。

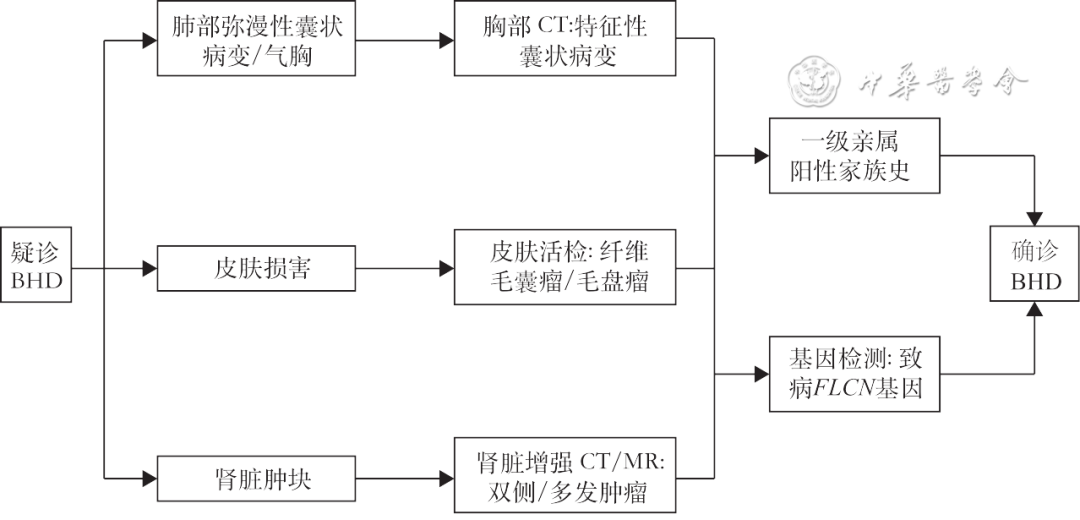
图5 伯特-霍格-杜布(BHD)综合征诊断流程图
【推荐意见4】对于临床及肺部影像表现疑诊BHD综合征患者,不建议首选肺组织病理活检。(投票专家43人;赞成43票,反对0票,弃权0票)。
BHD综合征肺组织活检病理上表现为肺内多发囊状病变,囊壁由肺泡壁上皮细胞构成,局灶见扩张的小血管,病变常紧邻胸膜下或叶间裂,缺乏诊断其他囊状病变的特征性病理形态,如朗格汉斯细胞、LAM细胞、轻链沉积等。由于BHD综合征病理组织学缺乏特异性改变,对于临床及影像表现疑诊BHD综合征患者,不建议首选肺组织病理活检。
【推荐意见5】BHD综合征重点需要与以下弥漫性肺部囊状病变鉴别,包括LAM、LIP及PLCH等。(投票专家43人;赞成43票,反对0票,弃权0票)。
【推荐意见6】BHD综合征临床罕见,常常涉及呼吸与危重症医学科、影像科、病理科、胸外科、泌尿外科、遗传学和皮肤科等多个学科,推荐开展多学科讨论,以提升该病诊断能力。(投票专家43人;赞成43票,反对0票,弃权0票)。
七、鉴别诊断
弥漫性囊状肺部病变(DCLD),其典型特征为双肺弥漫分布多个圆形或不规则形状、边缘有薄壁的肺部囊状病变的一组罕见疾病。根据潜在病因、病理生理学机制可分类为肿瘤性、先天性/遗传性、淋巴细胞增生性、感染性、吸烟相关性等,具体疾病包括淋巴管肌瘤病(LAM)、肺朗格汉斯细胞组织细胞增生症(PLCH)、淋巴细胞间质性肺炎(lymphocytic interstitial pneumonia,LIP)等,见图6。由于影像学类似(具体影像鉴别特征详见表2),常常容易混淆,临床上需要将BHD综合征与其他DCLD加以区分[44, 45, 46]。BHD综合征的临床表现涉及多个学科,需要呼吸与危重症医学科、影像科、病理科、胸外科、泌尿外科、遗传学和皮肤科等多学科讨论与合作,有助于提高该罕见病的诊疗能力,减少误诊和漏诊,并优化患者的全程管理[9]。
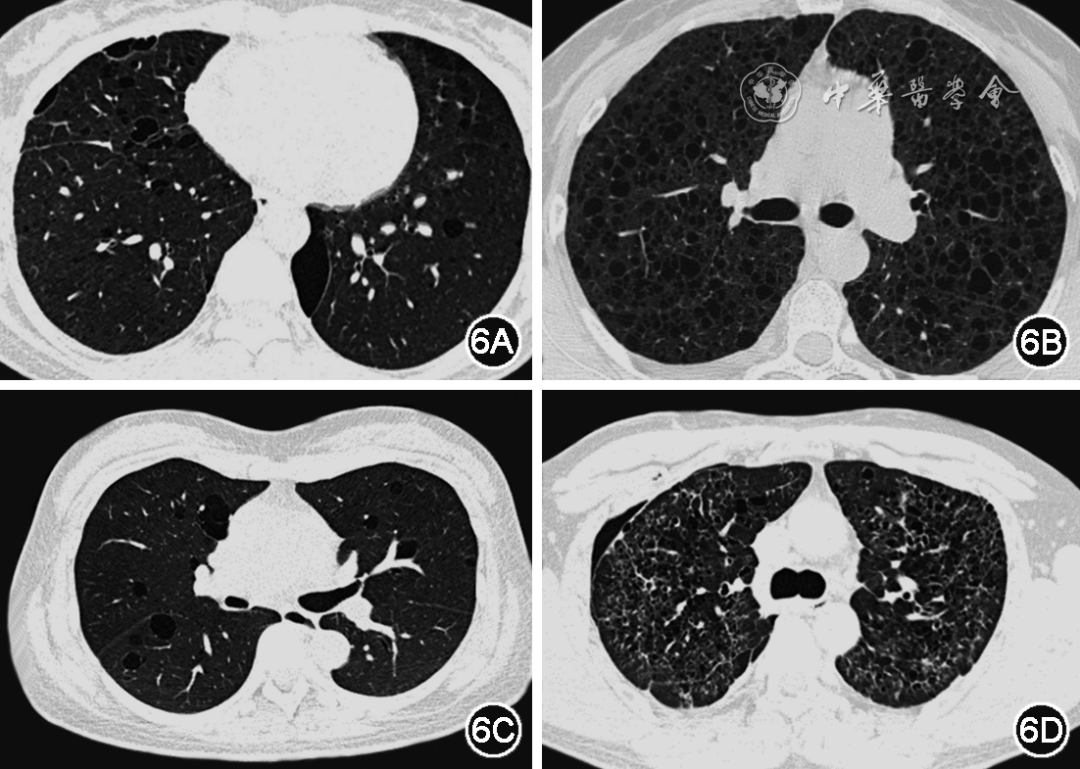
图6 弥漫性肺部囊状病变的CT影像表现,图6A为BHD综合征胸部CT可见双肺多发椭圆形或扁平状充气囊腔,多见于双肺基底部、纵隔旁、胸膜下分布;图6B为LAM胸部CT可见双肺均匀分布多发圆形囊腔;图6C为LIP胸部CT可见双肺散在分布肺囊腔,呈圆形或类圆形,内有组织结构或邻近血管;图6D为PLCH胸部CT可见双上肺多发不规则囊腔,大小不一,部分融合伴多发结节影,合并右侧气胸

1.淋巴管肌瘤病(LAM):LAM是一种以平滑肌样细胞异常增殖为特征、多器官受累的低度恶性肿瘤性疾病,主要发生于女性,其特征包括呼吸困难、双肺囊状病变,反复气胸、乳糜胸、腹腔淋巴结与腹膜后受累。LAM也常见于女性结节性硬化症(tuberous sclerosis complex,TSC)患者,即遗传型TSC-LAM。目前临床上多见为散发型LAM(S-LAM)。诊断LAM时需考虑:(1)详细采集个人史和家族史,了解是否有TSC或LAM的临床表现;(2)血清血管内皮生长因子-D(vascular endothelial growth factor D,VEGF-D)检测,约50%~70%的LAM患者中VEGF-D水平升高,当血清中VEGF-D水平>800 ng/L时,可辅助诊断LAM;(3)腹部增强CT或MR,可筛查是否存在腹膜后淋巴管肌瘤和血管平滑肌脂肪瘤;(4)经支气管镜肺活检,或胸腔镜手术肺活检获得病理诊断。
2.LIP:根据病因,LIP可分为特发性和继发性,临床上以继发性为主,主要继发于结缔组织病等,其中尤其以干燥综合征最为常见,约12%~46%的干燥综合征患者可出现肺部囊状病变。LIP可出现限制性通气功能障碍,伴弥散功能下降;通常根据病史、影像特征,部分患者风湿指标异常等有助于鉴别LIP与BHD综合征。
3. 肺朗格汉斯细胞组织细胞增生症(PLCH):PLCH是一种原因不明的罕见疾病,其特点是骨髓朗格汉斯细胞浸润至肺部和其他器官,LCH细胞可携带BRAF基因等基因变异。该病好发于年轻吸烟者,男性多见。PLCH临床特点表现为不同大小的结节性病变,浸润和破坏邻近组织相关结构,最常侵犯的器官包括骨骼、肺、皮肤、淋巴结和脑垂体。肺部受累表型为结节性病变可伴有弥漫性囊状改变,以中上肺野为主,且随着疾病加重,囊状病变可进展增多。
4. 其他类型的遗传性肾癌:遗传性肾癌种类较多,且大多亦具有双肾多发的特征,鉴别诊断时需结合其他器官系统的表现及基因检测结果。例如,对于VHL综合征相关肾癌,患者常伴有肾上腺嗜铬细胞瘤、胰腺囊腺瘤、中枢神经系统血管母细胞瘤等伴随病灶,同时在影像学中肾癌病灶常表现为肾透明细胞癌的特点,胚系基因检测可发现VHL基因突变。当需要与结节性硬化症相鉴别时,应关注肾脏病灶在MR中的表现,TSC相关肾肿瘤以血管平滑肌脂肪成分为主,免疫组化中HMB45的表达亦可提供鉴别价值。
【推荐意见7】BHD综合征患者应避免吸烟,推荐使用流感疫苗、肺炎球菌疫苗和新冠病毒疫苗预防感染。(投票专家43人;赞成43票,反对0票,弃权0票)。
【推荐意见8】BHD综合征并发气胸患者,在气胸未恢复前,不建议乘飞机出行。(投票专家43人;赞成42票,反对0票,弃权1票)。
八、治疗
BHD综合征的治疗目标包括积极处理气胸,降低其复发风险,定期筛查肾癌以改善其预后,开展家系排查及根据需要提供优生优育指导。
1.一般管理:尽管未有明确证据证明吸烟加重肺部囊状病变,仍强烈建议BHD综合征患者避免吸烟。推荐使用流感疫苗、肺炎球菌疫苗和新冠病毒疫苗预防感染。BHD综合征患者并发气胸的风险高,因此建议患者避免导致胸腔内压显著增高的活动,如举重物、潜水等运动等。对于近1个月内没有气胸的患者,可以行肺功能检查。BHD综合征患者通常可以乘坐飞机出行,但合并气胸(尚未治愈或1个月内有气胸史)的患者,不建议乘飞机出行。目前BHD综合征暂无对因治疗方法,临床上针对不同受累脏器的具体病变进行对症治疗,另外分子靶向药物治疗是未来研究的重要方向。
【推荐意见9】BHD综合征并发气胸患者,推荐尽早行胸膜固定术以降低气胸复发风险。(投票专家43人;赞成43票,反对0票,弃权0票)。
2.气胸:所有BHD综合征患者均应告知有发生气胸的风险,突发胸闷、胸痛等症状时应及时就医处理。由于BHD综合征患者气胸的复发率高达70%,发生气胸后推荐胸膜固定术降低其复发的风险。北京大学人民医院的回顾性研究显示外科手术治疗后BHD综合征气胸复发的风险仅为9.1%,较保守处理的53.1%复发率显著降低[10]。美国一项纳入104例BHD综合征的患者调查研究也显示类似的结果,胸膜固定术能降低同侧气胸复发风险约50%[47]。目前尚无前瞻性研究比较内科胸膜固定术和外科胸膜腔下胸膜固定术的疗效差异。近期日本一项纳入81例BHD综合征并发气胸的回顾性研究结果表明,采用氧化再生纤维素筛网胸膜固定术能显著降低气胸的复发率,其中部分胸膜固定术的5年复发率仅为12%,全胸膜固定术中位随访34个月均未见气胸复发(P=0.032)[48]。
【推荐意见10】BHD综合征并发皮肤纤维毛囊瘤或毛盘瘤患者,可选择观察或者局部治疗。(投票专家43人;赞成42票,反对0票,弃权1票)。
3.皮肤病变:BHD综合征患者皮肤损害常表现为良性肿瘤,常无严重临床症状,无需处理,建议定期随访。但出于美容角度,患者可采取电凝、激光、冷冻等局部治疗,通常效果良好,但存在复发情况。
【推荐意见11】BHD综合征并发肾脏肿瘤患者,肿瘤直径<1 cm,建议每年行腹部MR检查;肿瘤直径1~3 cm,建议每6个月行腹部MR检查,或者行消融手术;直径>3 cm的肾脏肿瘤,建议行保留肾单位的肾肿瘤局部切除术。(投票专家43人;赞成43票,反对0票,弃权0票)。
4.肾脏肿瘤:由于本病形成的肾脏肿瘤常累及双肾,且多数增长速度较慢,加之病情隐匿,因此尽早发现选择保留肾单位的肾肿瘤切除术非常重要,可减少多次手术带来的伤害。对于BHD综合征相关肾肿瘤手术,需要尽可能靠近肿瘤边缘,尽可能保留正常肾实质,若术前高度可疑为散发性嫌色细胞癌等,则切除更多的正常边缘。对于初诊即发现双侧肾脏肿瘤且均需手术干预者,建议简单、不需根治切除的一侧先行手术,术后3个月肾功能恢复后再行对侧手术[49]。最近一项法国研究报道6例BHD综合征经皮热消融术能有效清除共19个局部肾癌病灶,而且未发生肾功能恶化导致肾透析或者肾移植等不良事件,术后平均随访74个月未见复发[50]。因此,局部消融治疗也是可选择的治疗方式,但需要提前预判消融术后局部影像学的不典型变化对后续随访监测及外科手术难度的影响。肿瘤直径<1 cm,建议每年行腹部MR检查;肿瘤直径1~3 cm,建议每6个月行腹部MR检查,或者行消融手术;直径>3 cm的肾脏肿瘤,建议保留肾脏,行局部切除术;肾脏肿瘤切除后,建议每年行腹部MR检查直至5年,此后每2年行腹部MR检查,如果不能进行MR,可行腹部增强CT。
5.其他新型治疗方法:BHD综合征的动物模型显示mTOR功能调节异常,外敷的mTOR抑制剂西罗莫司已经成功用于治疗mTOR功能异常导致的结节性硬化症相关皮肤血管纤维瘤。因此,荷兰学者尝试用0.1%外用西罗莫司治疗BHD综合征的纤维毛囊瘤,但19例患者的随机对照双盲试验未能显示治疗有效的证据[51]。随着对BHD综合征认识和研究的深入,期待有更多新的治疗手段出现。
【推荐意见12】BHD综合征患肾癌风险显著增高,推荐终生定期筛查。(投票专家43人;赞成43票,反对0票,弃权0票)。
【推荐意见13】罹患BHD综合征的夫妻,建议在备孕期间进行孕产前遗传咨询,并与产前诊断专家共同评估遗传风险,并讨论孕期产前诊断的可行性。(投票专家43人;赞成43票,反对0票,弃权0票)。
【推荐意见14】BHD综合征患者家系成员应接受健康教育,推荐成年进行症状前FLCN基因检测,及时排除BHD综合征。(投票专家43人;赞成43票,反对0票,弃权 0票)。
九、患者随访和家系管理
BHD综合征尽管往往存在肺部囊状病变且容易发生气胸,但通常不影响肺功能,一般不需要每年复查胸部CT和肺功能检查。由于BHD综合征患者肾癌风险显著增高,因此建议对BHD综合征较早进行肾癌的筛查,可以从20岁开始,每3年进行一次肾脏肿瘤的筛查,为了减少放射线暴露,MR可作为首选的筛查手段[43]。此外,BHD综合征少见的临床表现包括大肠癌等恶性肿瘤[52]。因此在患者年度随访中,建议关注相关的症状和体征。
BHD综合征一般为成年发病且病情可以控制。对于BHD综合征患病夫妇,如果FLCN致病变异已明确,可以进行孕前的植入前遗传检测、产前诊断遗传咨询,明确检测方法的可行性和具体过程,在检测前签署知情同意书,这也是给予伦理心理咨询的最佳时间[53]。
BHD综合征为常染色体显性遗传病。通常情况下,患者父母之一也为BHD综合征患者;如果患者的双亲并非患者,则该患者可能为FLCN基因的新发变异导致。患者的同胞患病的可能性取决于患者的双亲之一是否为BHD综合征患者,即是否也携带FLCN基因变异。如果患者的双亲均正常,则患者的同胞患病可能性较低,但应高于一般群体,因为患者的父母也可能为生殖细胞嵌合;如果患者的双亲之一患病,则患者同胞有50%的可能患病。患者的子女罹患本病的概率是50%。BHD综合征患者家系成员,可进行症状前FLCN基因检测,及时排除BHD综合征。当先证者FLCN基因致病变异明确时,可直接针对该变异进行症状前基因检测(PCR一代测序或MLPA),以排除或明确BHD综合征的可能性。
【推荐意见15】BHD综合征临床诊治和全程管理水平仍有待提高,尤其是目前缺乏有效的治疗药物,建议开展多中心、前瞻性临床研究。(投票专家43人;赞成43票,反对0票,弃权0票)。
十、未来研究方向和展望
BHD综合征的漏诊和误诊情况并不少见,需要临床各科医生提高诊断的警觉性,特别是呼吸与危重症医学科、影像科、病理科、胸外科、皮肤科、泌尿外科等学科医生,须熟悉本病的临床表现、诊断流程和鉴别诊断。多学科讨论在BHD综合征诊治和管理中尤为重要。本共识有助于推进BHD综合征的临床诊治与全程管理,促进相关基础及转化研究。BHD综合征中国人群,有不同于欧美人群的临床表型和分子遗传特征,亟需进一步加强基础研究与临床探索,并规范指导临床评估和治疗。目前BHD综合征还缺乏有效的治疗药物,故该疾病的发病分子机制研究,特别是FLCN基因功能学研究和临床新治疗方法的探索需要进一步深入。最后,期待未来通过开展多中心的前瞻性临床研究,助力我国BHD综合征防治工作取得新突破。
伯特-霍格-杜布综合征诊治和管理中国共识(2023)专家组名单
顾问:钟南山(广州医科大学附属第一医院);张学(哈尔滨医科大学)
组长:胡晓文[中国科学技术大学附属第一医院(安徽省立医院)];刘杰(广州医科大学附属第一医院 国家呼吸医学中心)
执笔:胡晓文;刘杰;徐凯峰;田欣伦;欧阳若芸;易龙;韦炜
方法学专家:江梅(广州医科大学附属第一医院)
编写专家组(按照拼音字母排序):陈淮(广州医科大学附属第二医院);陈愉生(福建省立医院);董亮(山东第一医科大学第一附属医院);冯瑞娥(中国医学科学院北京协和医院);龚侃(北京大学第一医院);顾莹莹(广州医科大学附属第一医院);郝哲学(广州医科大学附属第一医院);胡晓文[中国科学技术大学附属第一医院(安徽省立医院)];贾楠、江梅(广州医科大学附属第一医院呼吸疾病国家重点实验室);金洪(四川大学华西医院);金美玲(复旦大学附属中山医院);林俊彦(广州国家实验室);刘杰(广州医科大学附属第一医院 国家呼吸医学中心);刘金丽[中国科学技术大学附属第一医院(安徽省立医院)];刘雅萍(中国医学科学院基础医学研究所);刘彦国(北京大学人民医院);陆国辉(南方医科大学珠江医院);欧阳若芸、彭红(中南大学湘雅二医院);田欣伦、王涛(中国医学科学院北京协和医院);韦炜、肖峻[中国科学技术大学附属第一医院(安徽省立医院)];肖永龙(南京大学鼓楼医院);谢敏(华中科技大学同济医学院附属同济医院);徐凯峰(中国医学科学院北京协和医院);徐文帅(清华大学附属长庚医院);杨燕丽(中国医学科学院北京协和医院);易龙(南京大学医学院江苏省医学分子技术重点实验室);张伟宏(中国医学科学院北京协和医院);张玉石(中国医学科学院北京协和医院);周希亚(中国医学科学院北京协和医院);朱成楚(温州医科大学附属台州医院);纵单单(中南大学湘雅二医院)
外审专家组名单(按照拼音字母排序):蔡后荣(南京大学鼓楼医院);代华平(中日友好医院);高占成(北京大学人民医院);瞿介明(上海交通大学医学院附属瑞金医院);徐金富(同济大学附属上海市肺科医院);徐作军(中国医学科学院北京协和医院);应可净(浙江大学医学院附属邵逸夫医院)
秘书:徐飞[中国科学技术大学附属第一医院(安徽省立医院)];盘颖新、黄虹元(广州医科大学附属第一医院)
参考文献(略)

作者:伯特-霍格-杜布综合征诊治和管理中国共识(2023)专家组 中国罕见病联盟呼吸病学分会 中华医学会呼吸病学分会 中国初级卫生保健基金会华南区呼吸罕见病专业委员会
通信作者:胡晓文,中国科学技术大学附属第一医院(安徽省立医院)呼吸与危重症医学科 中国科学技术大学附属第一医院罕见病诊疗中心;刘杰,广州医科大学附属第一医院呼吸与危重症医学科 国家呼吸医学中心 广州呼吸健康研究院 呼吸疾病国家重点实验室 国家呼吸系统疾病临床医学研究中心
引用本文: 伯特-霍格-杜布综合征诊治和管理中国共识(2023)专家组,中国罕见病联盟呼吸病学分会,中华医学会呼吸病学分会,等. 伯特-霍格-杜布综合征诊治和管理中国专家共识[J]. 中华结核和呼吸杂志, 2023, 46(9): 897-908. DOI: 10.3760/cma.j.cn112147-20230705-00362.
本文转载自订阅号「中华结核和呼吸杂志」(ID:cmjlung)
原链接戳:【诊疗方案】伯特-霍格-杜布综合征诊治和管理中国专家共识
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry