
 分享
分享
引言
本病例存在多器官、多系统病变,临床表现多样,而最终确诊为一种罕见的肺间质病变。求因过程中,应善于总结病例特征,尤其拎出一条主要特征进行层层剥茧分析,是最快得到明确指向的诊断思路。因此,提示我们面对这类疾病表现,要尽可能地用「一元论」断案,通过已知关键信息,分别考虑每个关键信息所包含的疾病,寻找交叉点,再进一步分析。在诊断方向较为明晰的前提下,再综合患者的病史、实验室检查及肺部CT特征性表现进行推敲、反证。本案中,吸烟、肺CT表现均为诊断的关键线索,而肺活检对于肺间质性疾病的诊断起到至关重要的作用。在治疗方面,我团队对此类病例有一些经验之谈,愿与同道分享,共同探讨。
咳嗽、多尿3年,颈部增粗2月……「双肺弥漫性病变查因,中枢性尿崩症」首次住院后仍有反复,气胸、颈部增粗接踵而至
患者是一名31岁的男子。主诉「咳嗽、多尿3年,颈部增粗2月」于2018年5月2日入住中南大学湘雅二院呼吸与危重症医学科。3年前(2015年),患者无明显诱因出现咳嗽,特点为阵发性连声咳,5-6阵/天,以干咳为主,剧烈咳嗽时痰中带血,伴烦渴、多尿,尿量每日可达5000ml以上,夜尿达6-7次。无发热,无乏力、盗汗,无脱发、口腔溃疡、关节痛、光过敏等不适。2015年12月,患者于外院就诊肺部CT提示双肺弥漫性大小囊性病变及小结节状影。头部MR提示垂体柄增粗。尿比重降低,禁水-加压素试验提示中枢性尿崩。诊断为「双肺弥漫性病变查因,中枢性尿崩症」,未明确诊断。予以止咳化痰、去氨加压素减轻尿崩等治疗。患者烦渴多尿症状好转,但仍有干咳。出院后患者仍有反复咳嗽。
2016年1月及2016年8月,患者2次无明显诱因突发胸痛、气促,当地医院胸片提示「气胸」,行胸腔闭式引流后逐渐缓解。2个月前,患者自觉颈部增粗,伴有右上腹痛,进食后明显,无吞咽困难、声音嘶哑,无恶心、呕吐、腹胀等不适。为进一步诊治,入住我科。
起病以来,患者精神、食欲、睡眠可,大便正常,小便次数增多,体重无明显改变。患者既往史:痛风病史4年,否认结核病及密切接触史,无药物过敏史。患者个人史:吸烟10余年,1包/天,已戒烟3年。未婚,家族史无特殊。
辅助检查情况如下:
1、2015年12月,患者于外院行肺部CT提示:双肺弥漫性大小囊性病变及小结节状影。尿比重降低,禁水-加压素试验提示中枢性尿崩。头部MR提示垂体柄增粗。
2、入院检查:
体格检查:体温:36.3℃;脉搏:98次/分;呼吸:20次/分;血压:118/81mmHg神清,体型中等,头皮及双侧腋窝多发浅表结节,突出皮肤,伴触痛,表面可见结痂,无分泌物。双侧颈部可触及肿大淋巴结约花生大小,左右各一颗,质地中等,活动可,无触痛。甲状腺Ⅲ度肿大,无血管杂音。双肺呼吸音粗,右肺下肺叶可闻及散在湿性罗音,无胸膜摩擦音。心、腹无阳性体征。骨关节及神经系统未见异常。指甲有纵行紫癜、纵向开槽。(图1)
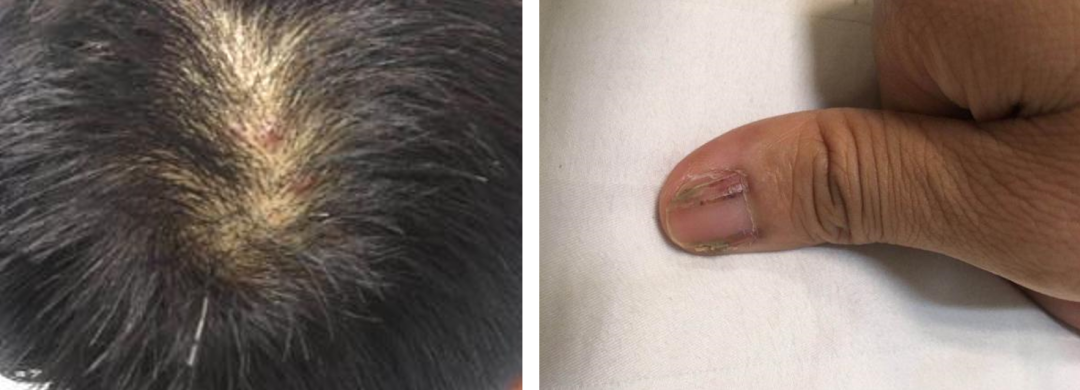
图1:皮肤改变:腋窝、头皮处皮肤毛囊炎改变,有结节。指甲:有纵行紫癜、纵向开槽,并有角化过度、甲剥离。
检验检查:
血气:PH:7.45,PCO2:38mmHg,PO2:83mmHg;
尿常规:尿比重1.002↓;
肝肾功能:谷草转氨酶85.9u/l↑,谷丙转氨酶84.0u/l↑,总胆红素20.6umol/l↑,直接胆红素10.4umol/l↑,尿酸512.9umol/l↑;乳酸脱氢酶305.7u/l↑;
血沉62mm/h↑
C反应蛋白71.30mg/l↑
G试验178.0pg/ml↑,GM试验0.19;
血常规、大便常规、电解质、血糖、血脂、凝血功能、正常。
甲状腺功能:T3:0.79ng/ml↓, T4:4.95ug/dL↓,TSH:27.81uIU/ml↑,甲状腺抗体:TG、TPO、TSHR抗体均正常。
肝炎全套、HIV、TP:阴性。血清铜兰蛋白:正常。
结缔组织全套:自免肝九项:抗ENA抗体-gp210抗体+;ANA、ENA、血管炎三项、ANCA、类风湿因子、抗CCP、抗磷脂抗体、补体均:正常。
皮质醇节律、ACTH节律:正常。
心电图:窦性心律,正常心电图。
心脏彩超:未见明显异常。
肺功能:肺弥散功能重度损害,重度混合型通气功能障碍。
肺部HRCT:双肺多发不规则囊样病变,纵隔多发稍肿大淋巴结,性质待定:不典型朗格汉斯组织细胞增生症?淋巴增生性疾病?其它?(图2)。
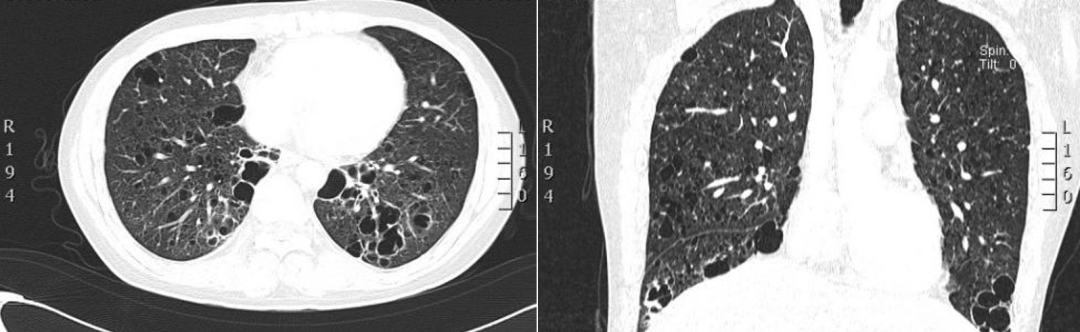
图2:肺部HRCT
进行颈部+全腹增强CT示:肝内多发低密度小结节灶,双侧甲状腺肿大(图3)。
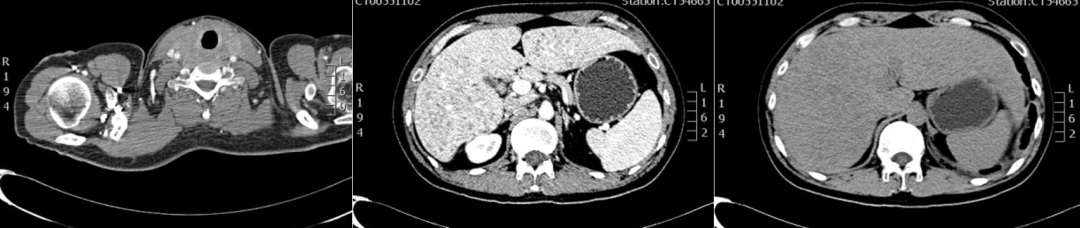
图3:颈部+全腹增强CT
团队首先对该患者的临床特点进行回顾:
1、症状:① 31岁青年男性;② 有吸烟史。③ 以干咳、多饮、多尿起病,期间多次出现自发性气胸;近期出现颈部增粗,右上腹痛。
2、体征:有皮疹、指甲改变;浅表淋巴结增大;甲状腺肿大。
3、辅助检查:① 实验室检查:血常规、血气均正常;肝功能出现转氨酶、胆红素升高;尿酸升高;② 甲状腺功能提示亚临床甲减,结缔组织检查-gp210抗体+,提示不能排除存在自身免疫性肝炎或原发性胆汁郁积性肝炎;③ 胸部CT:双肺多发不规则囊样病变;全腹CT:肝脏弥漫性病变;④ 外院完善禁水-加压素试验提示中枢性尿崩。头部MR提示垂体柄增粗。
总结病例特点:① 31岁青年男性;② 有吸烟史。③ 以干咳、多饮、多尿起病,期间多次出现自发性气胸;近期出现颈部增粗,右上腹痛。④ 有皮疹、指甲改变;浅表淋巴结增大;甲状腺肿大。⑤ 有尿崩症。⑥ 肺部CT提示双肺弥漫性大小囊性病变及小结节状影。
因此,可以围绕「双肺多发囊性病变、中枢型尿崩症、垂体柄增粗」三个特征进行详细分析。
思考题:求因可围绕三个特征进行,但着重应考虑以哪个特征为主要诊断思路?
面对诊断较为复杂的双肺囊性病变应作何考虑?……层层剥茧,诊断思路清晰明朗
目前可围绕三个特征进行分析,但考虑到其中双肺囊性病变较为复杂,诊断思路线路图如下:
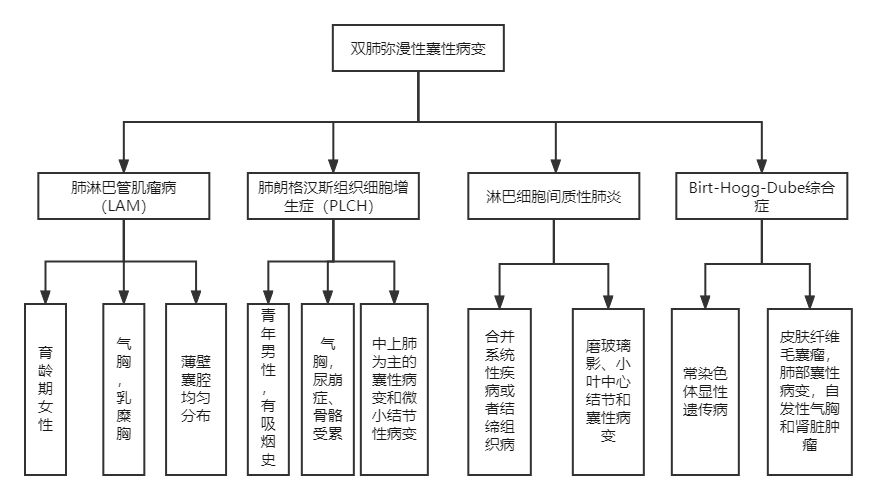
图4:双肺囊性病变诊断分析线路图
双肺弥漫性囊性病变的病因有哪些?我们首先根据弥漫性肺部囊性病变进行分类,主要病因有:
(1)肺淋巴管肌瘤病(LAM):该病多发于育龄期女性,反复气胸,常合并乳糜胸、乳糜腹、胸腹淋巴结增生,肾血管肌脂瘤。肺部CT表现为多发边界清楚的薄壁囊腔,均匀分布,一般无结节病变;
(2)肺朗格汉斯组织细胞增生症(PLCH):此病多发于青年男性,有吸烟史,反复气胸,常合并尿崩症、或骨骼受累。肺部CT表现为中上肺为主的囊性病变和微小结节性病变,后期囊腔融合,大小不一,性质不规则,结节逐渐消失;
(3)淋巴细胞间质性肺炎:此病通常会合并系统性疾病或者结缔组织病,如干燥综合征、类风关、系统性红斑狼疮等,特征性的影像表现包括磨玻璃影、小叶中心结节和囊性病变;
(4)Birt-Hogg-Dube综合征:BHD综合征是一种常染色体显性遗传病,临床特征为皮肤纤维毛囊瘤,肺部囊性病变,自发性气胸和肾脏肿瘤。其他少见病因包括:清链沉积病、滤泡性细支气管炎、恶性肿瘤转移灶(如肉瘤)、遗传性疾病(如马凡综合征、神经纤维瘤病Ⅰ型)。
中枢性尿崩症(CDI)的病因有哪些?该病主要分为三种类型:先天性CDI、获得性CDI和遗传性CDI。
(1)先天性中枢性尿崩:主要有家族性中枢性尿崩症、家族性垂体功能减退症以及先天性巨细胞病毒感染引起的尿崩症,占尿崩症的50%-60%;
(2)获得性中枢性尿崩:常见于:① 头颅外伤及垂体下丘脑手术:是CDI的常见病因。以脑垂体术后一过性CDI最常见。如手术造成正中隆突以上的垂体柄受损,则可导致永久性CDI。② 肿瘤:颅咽管瘤、垂体转移癌、垂体肉瘤、淋巴瘤等。③ 肉芽肿:结节病、朗格汉斯组织细胞增多症、类肉瘤、黄色瘤等。④ 感染性疾病:脑炎、脑膜炎、结核、梅毒、弓形体病等。⑤ 血管病变:动脉瘤、主动脉冠状动脉搭桥。⑥炎 症性:淋巴细胞性漏斗部神经垂体炎、肉芽肿病、红斑狼疮、硬皮病等;⑦ 化学毒物;⑧ 特发性;⑩ 其他:自身免疫性病变也可引起CDI,血清中存在抗AVP细胞抗体。
(3)遗传性中枢性尿崩症:可为X-连锁隐性、常染色体显性或常染色体隐性遗传。
这里要注意到,先天性中枢性尿崩症、特发性中枢性尿崩症以及自身免疫性中枢性尿崩症都是因为神经垂体系统本身病变所致,也称为原发性中枢性尿崩症;外伤、肿瘤、手术、感染、肉芽肿和血管病变所致的尿崩也称为继发性中枢性尿崩症。
垂体柄增粗的原因有哪些?该病可为肿瘤性、炎症性和先天性疾病三大类。肿瘤性疾病包括:生殖细胞肿瘤、颅咽管瘤和包含垂体腺瘤、垂体细胞瘤、星形细胞瘤、脑膜瘤、室管膜瘤等在内的一系列的颅内肿瘤,以及实体恶性肿瘤或血液系统肿瘤的垂体转移;炎症性疾病包括:自身免疫性垂体炎、朗格汉斯组织细胞增生症、神经系统结节病以及韦格纳肉芽肿等;先天性疾病:以Rathke囊肿为主。
思考题:综合分析后不难有重点考虑的诊断结果,但还需要完善哪些检查项目?
三个病因同指朗格汉斯细胞组织细胞增生症,如何反证?……以往常需开胸肺活检,该患采用冷冻肺活检(右侧)大大降低风险和费用
结合以上三个病因,实际上现在所有的答案都已共同指向了朗格汉斯细胞组织细胞增生症(LCH)。LCH是一组少见且病因未明的组织细胞增生性疾病,以大量朗格汉斯组织细胞增生、浸润和肉芽肿形成,导致器官功能障碍为特征。LCH可以累及全身任何器官或系统,因此临床表现多样。累及肺部主要表现为肺内囊泡和结节,累及骨主要表现为扁平椎、溶骨性改变,累及皮肤主要表现为囊泡、皮炎(主要在头皮、腋窝处)、结节,累及肝主要表现为黄疸、低蛋白血症,肝大及肝内多发小结节,累及垂体主要表现为垂体柄增粗、尿崩症,累及淋巴结引起淋巴结肿大,累及甲状腺主要表现为甲状腺肿大及功能异常等。
值得注意的是,肺朗格汉斯细胞组织细胞增生症(PLCH)是一种罕见的肺间质病变,可以仅有肺受累或者是系统性朗格汉斯细胞组织细胞增生症的肺部表现。好发年龄为20-40岁,约90%的成人PLCH患者有吸烟史或者被动吸烟史。大约2/3的PLCH患者在首诊时有症状,呼吸困难和干咳最为常见。大约15%患者发生气胸。
综合目前的情况,还应对患者进行哪些检测?(多选)
综合患者病史、实验室检查及肺部CT特征性表现,我们考虑其为朗格汉斯细胞组织细胞增生症,完善各系统评估。
全身骨X线、全身骨扫描:未见明显骨质破坏。
肝脏MRI平扫+增强:肝弥漫性病变:朗格汉斯细胞增生症?(图5)。
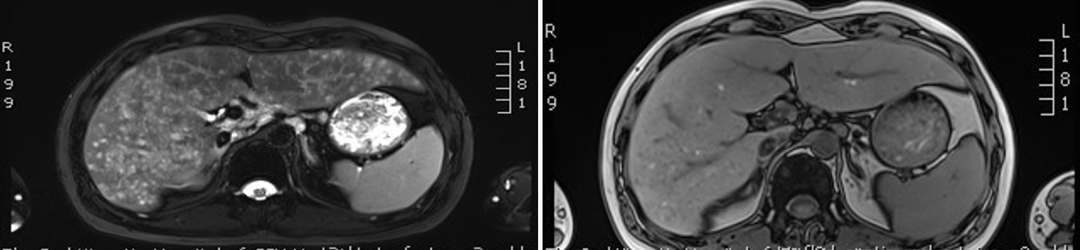
图5. 肝脏MRI平扫+增强
垂体MRI平扫+增强:部分空泡垂体。
检查结果均支持朗格汉斯细胞组织细胞增生症,支气管镜检查对PLCH的诊断具有一定作用,BALF中细胞免疫化学CDla朗格汉斯细胞阳性率>5%(正常值<1%)提示为PLCH。PLCH确诊依赖肺活检。诊断标准为:确诊:根据病理诊断,光镜符合朗格汉斯组织细胞表现,并且至少符合下列1条:CD207阳性、CDla阳性、电镜下见Birbeck颗粒。拟诊:根据病史及影像学拟诊,如肺CT表现为囊状病变和结节的吸烟患者。
为明确诊断,以往常需开胸肺活检,本例患者采用冷冻肺活检(右侧)的大大降低了风险和费用。
肺泡灌洗液细胞学:较多组织细胞、间皮细胞,少量淋巴细胞,未见典型恶性肿瘤细胞。肺泡灌洗液免疫组化:CD1a阳性细胞/组织细胞大于5%,不排除朗格汉斯组织细胞增多症可能,免疫组化:CD1a(+),CD68(+),CD27(+),Ki-67(20%+),S100(个别细胞+)、CD23(-)。冷冻肺活检病检回报:镜下结合免疫组化符合朗格汉斯组织细胞增多症,免疫组化:CD1a(+),S100(+),CD68(+),CD27(+),Ki-67(20%+)(图6)。
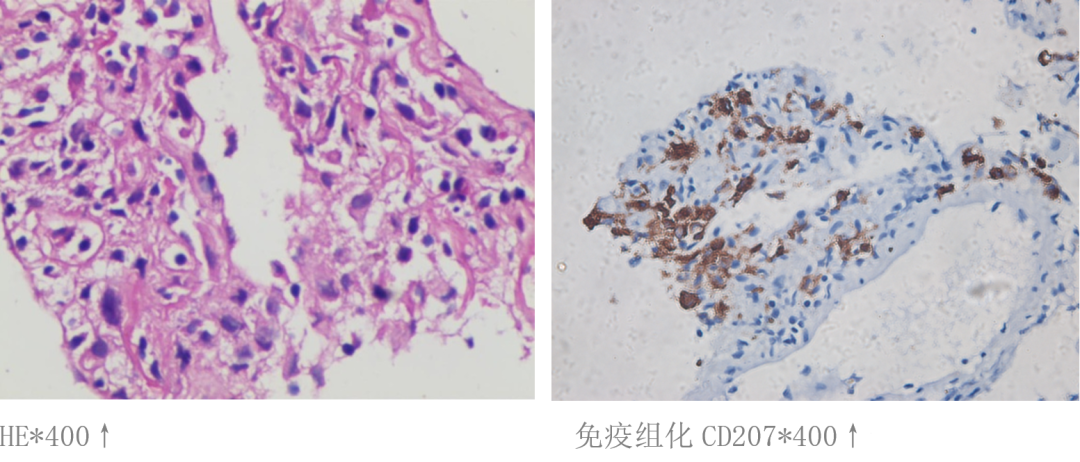
图6:肺活检病理结果及免疫组化
该患者最终诊断朗格汉斯组织细胞增生症(累及肺、肝、甲状腺、垂体、淋巴结、皮肤)继发性尿崩症。
治疗方面,予以戒烟,泼尼松片 60mg Qd 口服、去氨加压素 100ug Bid 口服控制尿崩症,辅以护肝、护胃、补钾、补钙等对症支持治疗,血液内科会诊建议转科行化疗。
患者行冷冻肺活检后10天,突发胸痛、气促,床旁X线提示左侧新发气胸。予以行床旁胸腔闭式引流术,引流后患者胸痛及气促减轻,建议患者气胸好转后到血液内科行进一步治疗。
最后诊断及诊断依据
最终诊断:朗格汉斯组织细胞增生症(累及肺、肝、甲状腺、垂体、淋巴结、皮肤)继发性尿崩症。依据如下:
1、症状:① 31岁青年男性;②有吸烟史。③以干咳、多饮、多尿起病,期间多次出现自发性气胸;近期出现颈部增粗,右上腹痛。
2、体征:有皮疹、指甲改变;浅表淋巴结增大;甲状腺肿大。
3、辅助检查:① 实验室检查:血常规、血气均正常;肝功能出现转氨酶、胆红素升高;尿酸升高;② 甲状腺功能提示亚临床甲减,结缔组织检查-gp210抗体+,提示不能排除存在自身免疫性肝炎或原发性胆汁郁积性肝炎;③ 胸部CT:双肺多发不规则囊样病变;全腹CT、肝脏MRI:肝脏弥漫性病变;④ 外院完善禁水-加压素试验提示中枢性尿崩。头部MRI提示垂体柄增粗;⑤ 垂体MRI平扫+增强:部分空泡垂体;⑥ 气管镜冷冻肺活检后,肺泡灌洗液结果及病理免疫组化符合朗格汉斯组织细胞增多症。
经验与体会
1、本例病例存在多器官、多系统病变,临床表现多样,尽可能用一元论,通过已知关键信息,分别考虑每个关键信息所包含的疾病,寻找交叉点,再进一步分析。
2、吸烟、肺CT表现为囊状病变和结节的是诊断关键线索,肺活检对于肺间质性疾病诊断至关重要。
3、成人PLCH治疗的关键部分是戒烟,戒烟可以部分甚至完全缓解病情。如果患者病情严重或者肺功能下降快,要考虑使用糖皮质激素,初始剂量推荐为泼尼松0.5-1.0mg/kg,逐月递减,连续服用6-12个月。严重的呼吸障碍和终末期患者行肺移植。对于成人多系统LCH缺乏统一的有效治疗方案。国内多参照欧洲组织学会推荐的6个周期泼尼松和(或)长春碱为基础的一线方案治疗多系统LCH。目前研究发现,在大约一半的患者的组织样本中观察到BRAFV600E的突变,对于此类患者BRAF激酶抑制剂可以作为靶向治疗而获益。
参考文献 (可上下滑动浏览)
[1] Steven H,Nancy LH,Stefano AP,et al.WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues[M].IARC press:Lyon,2008:358—360.
[2] Haupt R,Minkov M,Astigarraga I,et al.Langerhans cell histiocytosis(LCH):guidelines for diagnosis,clinical work-up,and treatment for patients till the age of 18 years[J].Pediatr Blood Cancer,2013,60(2):175-184
[3] Girschikofsky M,Arico M,Castillo D,et al.Management of adult patients with Langerhans cell histiocytosis:recommendations from an expert panel on behalf of Euro-Histio-Net[J].Orphanet J Rare Dis.2013,8:72.
[4] Gadner H,Minkov M,Grois N,et a1.Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis[J].Blood,2013,121(25):5006—5014.
[5] Kim HJ,Lee KS,Johkoh T,et al.成人肺朗格汉斯细胞组织细胞增生症的HRCT与病理学对照研究及其CT演变[J].国际医学放射学杂志,2011,34(5):495.
[6] Trotman-Dickenson B. Cystic lung disease: achieving a radiologic diagnosis.European journal of radiology.2014;83(1):39-46.
[7] 李业梅,俞小卫,杨明夏,等.成人朗格汉斯细胞组织细胞增多症二例[J].中华结核和呼吸杂志,2015,38(4):311-313.
[8] 唐华平, 李猛, 米娜, 等.成人肺朗格汉斯细胞组织细胞增多症二例[J].中华结核和呼吸杂志,2015,38( 5 ): 393-395.
[9] Narula G, Pradhan ND, Arora B, Banavali SD. Treatment of Langerhans
[10] cell histiocytosis with a modified risk-adapted protocol experience
[11] from a tertiary cancer institute in India. Pediatr Blood Cancer. 2018.
[12] https://doi.org/10.1002/pbc.27028.
专家介绍

罗红
教授,一级主任医师,博士生导师;中南大学湘雅二医院呼吸与危重症医学科主任、危重症亚专科主任;中华医学会呼吸病学分会呼吸危重症学组委员;中国医师协会内科医师分会委员;中国医师协会呼吸医师分会危重症医学工作委员会副主任委员;中国残疾人康复协会肺康复专委会常委兼ICU肺康复学组副组长;湖南省医师协会呼吸医师分会会长;湖南省医学会呼吸病学专业委员会副主任委员。

欧阳若芸
中南大学湘雅二医院呼吸与危重症医学科主任医师,教授,医学博士,博士生导师。2012年赴美国南加州大学医学中心访学1年。研究方向:睡眠呼吸障碍、肺部罕见病。担任中华医学会呼吸病学分会第十一届委员会睡眠呼吸障碍学组委员,中国医师协会呼吸医师分会睡眠呼吸障碍工作委员会委员,中国首届研究型医院学会罕见病分会理事,中国罕见病联盟呼吸病学第一届常务委员。主持国家级课题1项,参加国家级课题3项,主持省部级课题4项,获省级成果奖3项,发表专业学术论文60余篇,其中以第一作者或通讯作者发表SCI论文10余篇,Medline论文10余篇,获院医疗新技术奖2项。
贾玉楠
中南大学湘雅二医院呼吸科,助理研究员。参与国家科学自然基金青年项目1项、省自1项。中南大学湘雅二医院第二届中国医师节 “十佳住院医师”。主攻方向:呼吸重症、呼吸介入。
* 文章仅供医疗卫生相关从业者阅读参考
本文完
采写编辑:冬雪凝;责编:Jerry