
 分享
分享
目的:利用宏基因组二代测序(metagenomic next-generation sequencing,mNGS)技术分析肺部感染患者下呼吸道微生物特征,提高临床对于肺部感染患者病原学特征的认识。
方法:纳入在2020年8月—2021年10月期间,四川大学华西医院应用mNGS技术检测肺泡灌洗液微生物特征的840例疑似肺部感染患者,回顾性分析所有患者的下呼吸道微生物特征。
结果:共纳入840例患者,743例检出微生物阳性,其中细菌感染比例最高,占35.13%(261/743);细菌中鲍曼不动杆菌比例最高,占18.98% (141/743),其次是肺炎链球菌(14.13%、105/743),肺炎克雷伯菌(13.46%、100/743),屎肠球菌(12.11%、90/743)和结核分枝杆菌复合群(11.98%、89/743)。其中平均序列数最高的是鲍曼不动杆菌(2 607.48)。另外,也检出了一些特殊的病原体,如检出9例鹦鹉热衣原体。真菌感染主要为白色念珠菌(12.38%、92/743),耶氏肺孢子菌(9.02%、67/743)和烟曲霉(7.40%、55/743),其中耶氏肺孢子菌的平均序列数较高(141.86)。另外,也检出了一些特殊的病原体,如检出1例马尔尼菲篮状菌。病毒感染主要为人类β疱疹病毒5型(17.90%、133/743),人类γ疱疹病毒4型(17.36%、 129/743),人类β疱疹病毒7型(16.15%、120/743)和人类α疱疹病毒1型(13.59%、101/743),其中平均序列数最高的是人类α疱疹病毒1型(367.27)。寄生虫感染最少,只检出2例多房棘球绦虫,2例广州管圆线虫,2例屋尘螨和1例粉尘螨,以合并细菌和病毒感染为主。共有407例患者检出混合型感染,其中以病毒和细菌混合型感染最多(22.61%、168/743)。不同季节的微生物分布也存在一定的特征。秋冬季检出频数最多的是细菌(鲍曼不动杆菌),春夏季检出频数最多的是病毒(人类γ疱疹病毒4型)。
结论:肺部感染患者的下呼吸道微生物中,革兰阴性菌以鲍曼不动杆菌和肺炎克雷伯菌为主,革兰阳性菌以肺炎链球菌,屎肠球菌和结核分枝杆菌复合群为主;真菌以白色念珠菌,耶氏肺孢子菌和烟曲霉为主;病毒以人类β 疱疹病毒5型,人类γ疱疹病毒4型和人类β疱疹病毒7型为主;寄生虫感染很少,无明显特征;合并感染以细菌感染和细菌病毒混合感染为主;秋冬季和春夏季的微生物特征不同;另外,需要注意特殊的致病微生物,例如鹦鹉热衣原体和马尔尼菲篮状菌等。提示临床医生这些特征可以作为肺部感染病因诊断的参考与依据。
——————
呼吸系统感染是最常见的感染性疾病类型,众所周知,新冠肺炎是很严重的呼吸系统感染性疾病,因此防控呼吸系统感染性疾病意义重大[1]。因传统的检测方法在敏感性、特异性、时效性、信息量等方面存在局限,而且对于未知或者罕见的病原微生物,无法快速识别[2-4]。随着基因组学技术的发展,基于宏基因组二代测序技术(metagenomics next-generation sequencing,mNGS)直接针对样本中所有核酸进行无偏性测序,结合病原微生物数据库及特定算法,快速检测样本中含有的可能病原微生物(包括病毒、细菌、真菌、寄生虫),且无需特异性扩增,尤其适用于像新冠肺炎这种急危重症呼吸系统感染性疾病的诊断[5-6]。本研究回顾性地分析了四川大学华西医院2020 年8月—2021 年10 月采用mNGS技术诊断的840 例疑似肺部感染患者的肺泡灌洗液标本中的病原微生物数据,旨在分析肺部感染患者的临床感染微生物特征,为此类患者的早期诊断提供参考依据。
一、资料与方法
1、样本的基本信息
本研究纳入2020 年8月—2021年10 月四川大学华西医院采用mNGS 技术诊断的840例疑似肺部感染患者。其中,43 例为门诊患者,797 例住院患者;年龄14~95 岁,235例大于65岁;男542例,女298 例。鉴于收集的数据覆盖了一年多的时间段,对不同季节的微生物分布也进行了分析。根据成都的气候,定义秋冬季为9月—次年 2月,春夏季为3 月—8 月。
2、病原学mNGS检测及临床解读
840例患者均采集肺泡灌洗液标本进行病原微生物检测,采样步骤遵循以下标准[7]。
(1)样本处理和DNA提取:肺泡灌洗液经玻璃珠混合震荡后取 0.5~3 mL,后加入7.2 μL溶壁酶(RT410-TA, TIANGEN BIOTECH,北京)进行酶破壁反应,混合震荡后取300 μL样本,并按照TIANamp Micro DNAKit试剂盒(DP316, TIANGENBIOTECH,北京)说明书提取DNA(用作DNA文库构建)。
(2)文库构建和测序:对提取好的核酸进行酶切片段化、末端修复、接头连接及聚合酶链反应进行文库构建,使用 Agilent2100 Bioanalyzer质控文库,质控文库片段大小为200~300 bp左右,使用QubitdsDNAHS AssayKit(Thermo Fisher Scientific Inc.)质控DNA文库浓度,按照检测后的浓度将构建好的文库按照等质量进行pooling,将pooling混合后的文库经环化形成单链环形结构。再经滚环复制形成DNA纳米球,然后加载至测序芯片,采用MGI2000 进行高通量mNGS测序[8]。
(3)数据分析:测序所得原始结果首选去除质量低和接头污染的数据,过滤后的数据通过对比BWA(http://bio-bwasourceforgenet/)去除人参考基因组序列。剩下的数据除去低复杂度序列,然后比对BGI微生物参考数据库PMDB(包括6 350 种细菌、1 064种真菌、4 945种病毒、234 种寄生虫),将比对后的数据按照细菌、真菌、病毒、寄生虫、支原体/衣原体等进行分类,得到初步的结果。
(4)数据解读:结合阴控对照样本的检出排除污染和不可信的结果,所有物种的通用排除标准如下:① 该物种的每百万有效测序序列数(reads per million,RPM)<3 倍阴控样本中该物种的RPM;② 常见的已知污染物种;③ 种水平序列数<3;④ 该物种比对到参考基因组的分布不均匀,因为分布太集中,可能只是某个区域的随机片段,并不是真实序列;⑤ 文献中报道的呼吸道中常见定植菌;⑥ 同批次中其他样本存在该物种的强阳检出(序列数大于10 000,或者序列数远远大于其他所有样本),结合样本提取顺序排除;⑦ 常见的环境物种,不致病的物种。排除污染和不可信的结果后,由于细菌检出物种很多,再按照属水平的序列数进行排序,一般物种只考虑排名前10 的属,每个属中只考虑序列数排名前2 的种;对于致病性很强的常见物种(比如肺炎克雷伯菌),则不受该排名的限制,只要满足上述的排除标准就保留;真菌和病毒,只要满足上述的排除标准就保留;寄生虫由于基因组序列比较大,而且跟人的参考基因组相似度更高,除了满足上述的排除标准,还需要满足序列数>10。并且,结合患者的临床提示,选择最可信的物种,出具初步的检测报告。然后,临床医师结合传统检测和影像学检查等结果,出具最终的检测报告。本研究中纳入的数据为最终检测报告中的微生物数据。
在进行细菌分布特征分析时,由于不同微生物所获取的片段数差异,相对测序丰度低的细菌通过比对仅能确定至属,因此,对于结核分枝杆菌复合群中的物种,全部统一到属水平,而其他物种全部统一到种水平。
需要强调的是,临床医生需要依据患者病情、临床表现、影像学检查、经验,甚至多学科会诊,全面分析mNGS 结果,最终确定致病病原体[9]。
3、统计学方法
为了排除测序数据量对结果的影响,本研究中纳入的840例样本的微生物序列数统一采用RPM,然后再进行比较。使用R 4.0.3 统计学软件进行数据分析处理。采用Wilcoxon秩和检验分析两组样本之间的差异,P<0.05 为差异有统计学意义。
二、结果
1、总体微生物分布特征
840例患者的测序结果中,微生物阳性患者有743例(占88.45%);其中细菌感染阳性比例显著高于病毒或真菌感染者;其中743例测序阳性患者中检出频数最多的20 个微生物分布见图1。鲍曼不动杆菌比例最高,占18.98% (141/743),其次为人类β疱疹病毒5型(17.90%、133/743),人类γ疱疹病毒4型 (17.36%、129/743),人类β疱疹病毒7型(16.15%、120/743),肺炎链球菌(14.13%、 105/743),人类α疱疹病毒1 型(13.59%、 101/743),肺炎克雷伯菌(13.46%、100/743),白色念珠菌(12.38%、92/743),屎肠球菌(12.11%、 90/743),结核分枝杆菌复合群(11.98%、89/743),微小微单胞菌(9.83%、73/743),铜绿假单胞菌(9.29%、69/743),耶氏肺孢子菌(9.02%、67/743),烟曲霉(7.40%、55/743),嗜麦芽窄食单胞菌(6.33%、47/743),细环病毒(6.06%、45/743),星座链球菌(5.52%、41/743),痰液嗜血杆菌(4.58%、 34/743),副溶血嗜血杆菌(4.31%、32/743),热带念珠菌(4.04%、30/743)。

图1:743例测序阳性患者中检出最多的20个物种分布图
2、细菌分布特征
由于细菌的检出物种比较多,只展示频数最高的30 个细菌分布见表1。结果发现,检出频数最多的5个细菌为:鲍曼不动杆菌比例最高,占18.98%(141/743),其次是肺炎链球菌(14.13%、105/743),肺炎克雷伯菌(13.46%、100/743),屎肠球菌(12.11%、90/743)和结核分枝杆菌复合群(11.98%、 89/743)。其中,鲍曼不动杆菌和肺炎克雷伯菌的平均序列数也是排名最高的,分别为2 607.48 和250.21。另外,也检出了一些特殊的病原体,例如检出9例鹦鹉热衣原体。

3、真菌分布特征
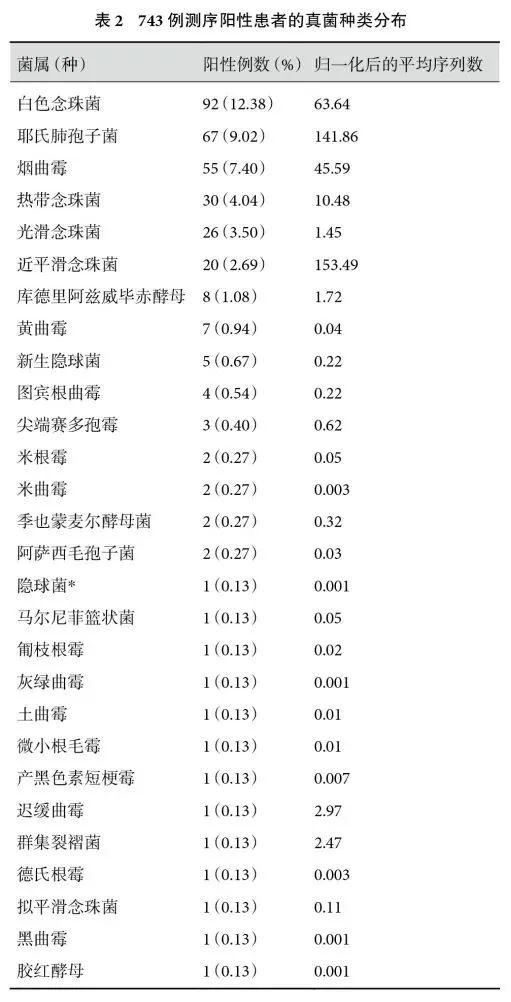
检出真菌的种类明显少于细菌,一共检出28 个物种,所有检出真菌的分布见表2。结果发现,检出频数最多的3 个真菌为:白色念珠菌(12.38%、92/743),耶氏肺孢子菌(9.02%、67/743)和烟曲霉(7.40%、55/743),这3个真菌的平均序列数也排名靠前,分别为白色念珠菌(63.64),耶氏肺孢子菌(141.86),烟曲霉(45.59)。另外,隐球菌检出6例,根(毛)霉检出5 例。值得一提的是,也检出了一些特殊的病原体,例如检出1 例马尔尼菲篮状菌,该病例是人类免疫缺陷病毒感染患者。
4、病毒分布特征
检出病毒的种类明显少于细菌,病毒统一到种水平,一共检出22个物种,所有检出病毒的分布见表3。检出频数最多的4 个病毒为:人类β疱疹病毒5型(17.90%、133/743),人类γ 疱疹病毒4型(17.36%、129/743),人类β疱疹病毒7型(16.15%、 120/743)和人类α 疱疹病毒1型(13.59%、 101/743),其中平均序列数最高的是人类α疱疹病毒1型(367.27),其次是人类β疱疹病毒5型(51.43)。另外,也检出了人类哺乳动物腺病毒 D组和人类哺乳动物腺病毒C型各1 例。

5、寄生虫分布特征
寄生虫检出种类很少,一共只检出4 个物种,所有检出物种见表4。具体为:检出2 例多房棘球绦虫,2例广州管圆线虫,2例屋尘螨和1 例粉尘螨。平均序列数分别为多房棘球绦虫(540.5),屋尘螨(107),粉尘螨(104)和广州管圆线虫(16)。值得一提的是本研究中纳入的数据表明寄生虫并不单独作为致病病原体,都是以合并细菌或者病毒感染的形式存在。提示临床,肺部感染患者也要留 意寄生虫导致的合并感染。

6、测序阳性患者的感染类型
对743例测序阳性患者的检出微生物物种,按照细菌,真菌,病毒和寄生虫这4 种进行分类,统计结果见表5。混合感染定义为检出2 种或以上分类的感染。结果发现,非混合感染中,细菌感染的比例最高(35.13%、261/743),混合感染中,细菌+病毒混合感染的比例最高(22.61%、168/743),其次是细菌+真菌+病毒混合感染(16.02%、 119/743),细菌+真菌(10.63%、79/743)和真菌+病毒(4.58%、34/743),而细菌+病毒+寄生虫以及病 毒+寄生虫的检出率较低(仅占0.81%和0.13%)。

7、不同季节微生物分布特征
不同季节的微生物分布也存在一定的特征。743例阳性样本包括343 例秋冬季样本和400例春夏季样本。首先,统计不同季节中检出最多的20 个物种做分布图(图2)。结果发现,秋冬季检出频数最多的是细菌(鲍曼不动杆菌),春夏季检出频数最多的是病毒(人类γ疱疹病毒4 型)。其次,为了比较秋冬季和春夏季这些检出物种的序列数,对以上两个图中的物种取并集得到24 个物种,统计这24 个物种的序列数(表6)。结果发现,有16 个物种的平均序列数,春夏季的检出要高于秋冬季的。并且,肺炎克雷伯菌(P = 0.024), 流感嗜血杆菌(P = 0.026), 脓肿分枝杆菌(P < 0.001), 痰液嗜血杆菌(P = 0.004), 微小微单胞菌(P < 0.001), 耶氏肺孢子菌(P = 0.015), 人类β疱疹病毒5型(P = 0.002),人类β疱疹病毒7型(P = 0.002)和细环病毒(P = 0.001)的平均序列数,在春夏季和秋冬季的比较中,为差异有统计学意义。

图2:743例测序阳性患者在不同季节中检出最多的20个物种分布图
a. 343例秋冬季阳性样本中检出最多的20个物种分布图;b. 400例春夏季阳性样本中检出最多的20个物种分布图。

三、讨论
肺部感染是发病率和死亡率最高的感染性疾病[10-14],给社会和个人造成了极其承重的疾病负担。由于造成该类感染的病原体存在多样性,多数医生在无法进行准确的病原学诊断前常采用经验治疗[15],但是经验性治疗的效果往往不佳。因此,快速且准确地找到该类感染的病原体,给予针对性的治疗,是迫切需要解决的问题[16]。mNGS可以进行无偏性测序,无需培养,病原体覆盖度广,灵敏度高,有利于鉴定不明病原体和混合感染[5-6]。本研究采用mNGS 技术系统性地分析了疑似肺部感染患者的下呼吸道微生物特征,对该类感染患者的病原诊断和精准治疗具有重要的临床意义。
本研究分析了840例疑似肺部感染患者的下呼吸道微生物特征,结果发现840例患者的测序结果中,微生物阳性患者有743例(88.45%);其中检出频数最多的5个细菌为:鲍曼不动杆菌比例最高,占18.98% (141/743),其次是肺炎链球菌(14.13%、 105/743),肺炎克雷伯菌(13.46%、100/743),屎肠球菌(12.11%、90/743)和结核分枝杆菌复合群(11.98%、89/743)。其中,鲍曼不动杆菌和肺炎克雷伯菌的平均序列数也是排名最高的,分别为2 607.48和250.21。即革兰阴性菌以鲍曼不动杆菌和肺炎克雷伯菌为主,革兰阳性菌以肺炎链球菌,屎肠球菌和结核分枝杆菌复合群为主;细菌谱特征与之前的结论基本一致[17];另外,检出频数最多的3个真菌为:白色念珠菌(12.38%、92/743),耶氏肺孢子菌(9.02%、67/743),烟曲霉(7.40%、 55/743),这3 个真菌的平均序列数也排名靠前,分别为白色念珠菌(63.64),耶氏肺孢子菌(141.86),烟曲霉(45.59)。即真菌谱特征以耶氏肺孢子菌、烟曲霉菌和白色念珠菌为主[18],而隐球菌、毛霉菌和地方性真菌检出较少但也值得关注。另外,检出频数最多的4个病毒为:人类β疱疹病毒5 型(17.90%、133/743),人类γ疱疹病毒4型(17.36%、 129/743),人类β疱疹病毒7 型(16.15%、120/743)和人类α疱疹病毒1型(13.59%、101/743),其中平均序列数最高的是人类α疱疹病毒1型(367.27),其次是人类β疱疹病毒5型(51.43)。即病毒的特征以人类β疱疹病毒5型,人类γ疱疹病毒4型和人类β疱疹病毒7型为主[12-13],该结果进一步证实,人类β疱疹病毒5型、人类γ疱疹病毒4型、和人类β疱疹病毒7型与肺部感染关系密切[12-13]。同时,也需要关注其他呼吸道病毒,比如人类哺乳动物腺病毒。而由寄生虫导致的肺部感染病例非常少,没有明显的特征;寄生虫并不单独作为致病病原体,都是以合并细菌或者病毒感染的形式存在。值得一提的是,非混合感染中,细菌感染的比例最高(35.13%、261/743),而混合感染中,细菌+病毒混合感染的比例最高(22.61%、168/743),其次是细菌+真菌+病毒混合感染(16.02%、119/743),细菌+真菌(10.63%、79/743)和真菌+病毒(4.58%、 34/743),而细菌+病毒+寄生虫以及病毒+寄生虫的检出率较低(仅占0.81% 和0.13%);说明mNGS在混合感染的检测方面有明显的优势,因为对于混合感染的检测是传统检测方法难以逾越的障碍。提示临床医生,这些特征可以为诊断此类肺部感染患者的病因提供一定的参考依据。
其次,本研究发现不同季节的微生物分布也存在一定的特征。例如,秋冬季检出频数最多的是细菌(鲍曼不动杆菌),春夏季检出频数最多的是病毒(人类γ疱疹病毒4 型),而且有9个物种的平均序列数在两组的比较中是差异显著的(P<0.05),表明不同季节的肺部微生物存在不同的特征[19],提示临床医生,不同季节的肺部感染病因可能存在差异,要针对性地治疗。
更重要的是,对于未知病原体的检测也是传统检测方法的不足之处。mNGS对于未知或者罕见的病原微生物的快速识别有明显的优势,该技术将为感染性疾病的诊断带来新的突破口[20],值得进一步推广和应用。本研究通过mNGS技术,测到了一些不常见的特殊病原体,例如鹦鹉热衣原体和马尔尼菲篮状菌等。鹦鹉热衣原体肺炎是一种动物源性感染性疾病,具有一定的临床症状、实验室检查和影像学特点,采用mNGS技术快速诊断并给予合适的治疗能明显缩短病程并改善预后[10]。据文献报道,马尔尼菲篮状菌感染已经发展成为艾滋病患者常见的机会性感染之一,提示患有艾滋病免疫功能低下的患者,要留意马尔尼菲篮状菌的合并感染[11]。提示临床医生,在遇到不明原因的肺部感染患者时,不能排除这些非典型的致病病原体,可以考虑采用mNGS技术进行辅助诊断。
但是,因mNGS 高度的敏感性,且临床样本采集及实验室测序诸多环节中均存在潜在的污染可能,其结果可能存在一定的假阳性,因此, mNGS 的数据解读是该技术中的一大难点,需要不断完善相关的标准和共识。另外,mNGS技术在实验的过程中也有待完善的地方,例如在样本采集过程中需要严格执行无菌化操作,在实验过程中需要保持实验室操作流程标准化。只有不断完善这些要点,才能使mNGS技术在感染性疾病的诊断中发挥更好的作用[21]。同时,临床医生也需要以科学严谨的态度,采用合理的方式将mNGS应用于临床实践。
本研究也存在一定局限性:本研究中纳入的数据为mNGS 检测的检测报告中提示的致病病原体,并不涉及随访数据,这些微生物是否全部都被确定为致病病原体,以及根据这些病原体的治疗效果,有待进一步确认。我们将在未来的研究中,纳入相关的数据,进行更多更深入的研究。
综上所述,本研究通过分析840 例疑似肺部感染患者的肺泡灌洗液标本中的微生物数据特征,发现肺部感染患者的下呼吸道微生物中,革兰阴性菌以鲍曼不动杆菌和肺炎克雷伯菌为主,革兰阳性菌以肺炎链球菌,屎肠球菌和结核分枝杆菌复合群为主;真菌以白色念珠菌,耶氏肺孢子菌和烟曲霉为主;病毒以人类β疱疹病毒5型,人类γ 疱疹病毒4型和人类β疱疹病毒7 型为主;寄生虫感染很少,无明显特征;合并感染以细菌感染和细菌病毒混合感染为主;秋冬季和春夏季的微生物特征不同;另外,存在特殊致病病原体,例如鹦鹉热衣原体和马尔尼菲篮状菌等。这些特征可以为此类肺部感染患者的病因诊断提供一定的参考依据,有助于早期精准地进行临床干预。
利益冲突:本研究不涉及任何利益冲突。
参考文献略。
引用本文:陆思芬,周永召,王刚,王婧,江娟,邓竹君,张文庚,李为民. 基于宏基因组二代测序技术的840 例疑似肺部感染患者下呼吸道微生物特征分析. 中国呼吸与危重监护杂志, 2022, 21(6): 403-411. doi: 10.7507/1671-6205.202112019
本文转载自公众号「中国呼吸与危重监护杂志」
原链接戳:基于宏基因组二代测序技术的840例疑似肺部感染患者下呼吸道微生物特征分析