
 分享
分享
推荐语
近日,国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布了《第一批罕见病目录》,共收录121种疾病,其中多种为呼吸疾病,为此《呼吸界》特约专家对此逐一进行分析解读,敬请关注。
2018年5月31日上午,中日医院特需门诊,一位来自云南的年轻母亲在剖宫产术后半年出现活动后气短、爬2层楼都困难的症状,当地医院超声显示肺动脉压力75mmHg,医院诊断为「特发性肺动脉高压」。
这位年轻患者一进诊室,就控制不住自己的眼泪,不停地问我:「我是不是真的得了原发性肺动脉高压?我是不是活不了几年了?我这个病还有希望吗?」「能告诉我还能陪孩子几年吗?我只希望能够有足够的时间多陪陪孩子。」我用了将近半小时的时间,向她和她的丈夫做了详尽的解释,并建议她住院进一步系统检查。我不知道能给她带来多少希望,但还是希望通过团队的努力,给她准确的评估,制定相应的方案,给这个妈妈,给这个家庭和他们的孩子带去曙光。
特发性肺动脉高压近年来在国内外医学界备受关注。2018年5月11日,国家卫健委、科技部、工信部、国家药监局、国家中医药管理局五大部门联合下发《关于公布第一批罕见病目录的通知》,特发性肺动脉高压被列入第一批罕见病。我们将与大家一起走近肺动脉高压,认识并了解特发性肺动脉高压。
问题一:什么是特发性肺动脉高压?
肺动脉高压(pulmonary hypertension)是由多种已知或未知原因引起的肺动脉压异常升高的一种病理生理状态,血流动力学诊断标准为:在海平面、静息状态下,右心导管测量平均肺动脉压(mean pulmonary artery pressure,mPAP)≥25mmHg。
肺动脉高压的原因众多,发病率不低,特发性肺动脉高压(idiopathic pulmonary arterial hypertension,IPAH)是指原因不明的一类肺动脉高压,发病率低,属于罕见病的范畴,但诊断、治疗难度大,多学科合作尤其重要。
欧洲资料显示,成年人IPAH的患病率约为5.9/100万人。目前我国尚无发病率的确切统计资料。1981年美国国立卫生院第一次注册研究数据显示IPAH的平均患病年龄为36岁。
既往认为女性更容易患本病,但近年来的研究有不同的结果,性别差异在本病中无明显体现,尤其是老年患者。我国学者研究表明,仅接受传统药物治疗,IPAH与家族性肺动脉高压患者1年、3年、5年的生存率分别为68%、38.9%、20.8%,接受肺动脉高压靶向药物,患者1年、3年、5年的生存率分别为84.1%、73.7%、70.6%。
特发性肺动脉高压主要影响远端肺动脉(<500μm),表现为中膜增厚,内膜增生和纤维化改变,外膜增厚合并轻-中度炎症细胞浸润和淋巴细胞增生,复合病变(丛状、扩张病变),血栓形成。
肺动脉高压的发病机制
(引自Bellerophon Therapeutics)
问题二、如何早期识别和诊断特发性肺动脉高压?
特发性肺动脉高压患者临床症状无特异性,可有乏力、呼吸困难、干咳和晕厥,通常劳累后加重,重症患者静息状态亦可出现症状。
体格检查可发现胸骨旁抬举样搏动,肺动脉瓣听诊区第二心音亢进,右心室可闻及第三心音,三尖瓣听诊区收缩期反流性杂音及肺动脉瓣听诊区舒张期反流性杂音。病情进展合并右心衰竭时,可出现颈静脉怒张、肝脏肿大、腹水、四肢水肿等。
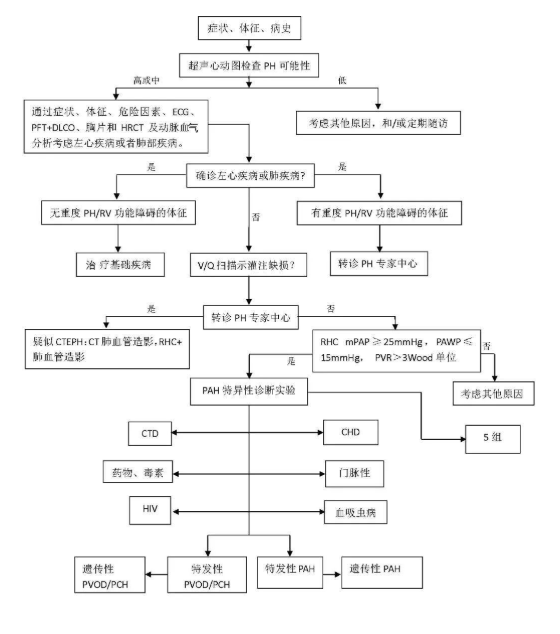
特发性肺动脉高压的诊断需要综合多种辅助检查, 其中超声心动图、右心导管检查尤为重要。
超声心动图
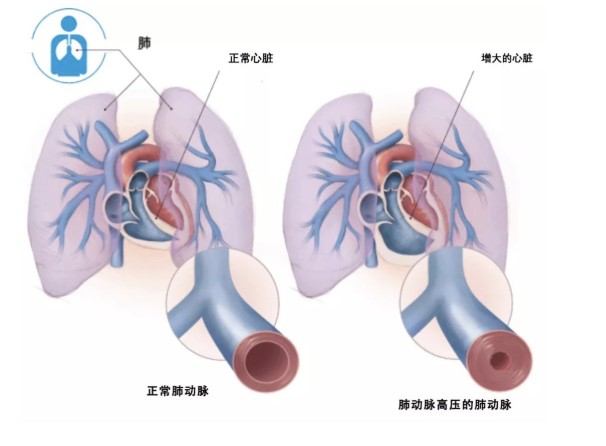
经胸超声心动图可用来估测肺动脉压力,并评估肺动脉高压对心脏的影响,是筛查肺动脉高压最重要的无创性检查方法,多普勒超声心动图估测三尖瓣峰值流速>3.4m/s或肺动脉收缩压>50mmHg将被诊断为肺动脉高压 【表1】。
【表1】超声心动图在肺动脉高压中的评估及临床建议:
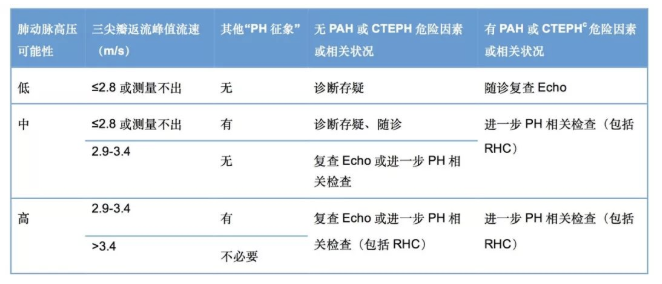
* 其他PH征象:右心室、肺动脉、下腔静脉和右心房的超声心动图征象。
右心导管检查
右心导管检查是确定肺动脉高压的金标准检查,可直接测量肺动脉压力,并测定心排出量,计算肺血管阻力,确定有无左向右分流等,有助于制定治疗策略。
急性血管反应试验
急性血管反应试验(acute vasoreactivity test)是评价肺血管对短效血管扩张剂的反应性,目的是筛选出对口服钙通道阻滞剂可能有效的患者。急性肺血管反应试验阳性标准为mPAP下降≥10mmHg,且mPAP下降到≤40mmHg,同时心排出量增加或保持不变。
其他
特发性肺动脉高压的发病与BMPR2、ACVRL1和ENG等基因突变相关,相关基因的检测对于诊断及认识疾病发病机制均有重要意义。其他检查包括心电图、胸片、肺功能、血气分析、肺通气/灌注核素扫描、胸部高分辨CT、CT肺血管造影、心脏核磁、血液化验、免疫学检查和腹部B超等均有诊断与鉴别诊断意义。
多普勒超声心动图估测三尖瓣峰值流速>3.4m/s或肺动脉收缩压>50mmHg可被诊断为肺动脉高压,肺动脉高压的确诊标准是右心导管检查测定平均肺动脉压≥25mmHg。但IPAH属于排除性诊断,必须在除外各种引起肺动脉高压的病因后方可作出诊断。
具体诊断流程见下表:
肺动脉高压患者的综合管理
(引自CHEST Foundation)
问题三、如何早期治疗和管理特发性肺动脉高压?
治疗策略包括:(1)初始治疗及支持治疗。(2)急性血管反应试验阳性患者给予高剂量CCB类药物治疗,急性血管反应试验阴性患者给予靶向药物治疗。(3)对于治疗反应不佳的患者,联合药物治疗及肺移植。需要强调的是,IPAH的治疗一定是在吸氧、利尿、纠治贫血等基础之上的全面治疗。
初始治疗
建议育龄期女性患者避孕;及时接种流感及肺炎链球菌注射疫苗;给予患者社会心理支持;体力下降患者在药物治疗的基础上进行必要的康复训练;WHO功能分级III-IV级和动脉氧分压持续低于8 kPa(60 mmHg)的患者建议进行氧疗;如需要进行手术,首选硬膜外麻醉而非全麻。
支持治疗
1、口服抗凝药物
IPAH患者的尸检显示了血管内原位血栓形成的高患病率,凝血及纤溶途径异常也有报告,静脉血栓栓塞症的非特异高危因素包括心衰、制动,以上都是进行口服抗凝药物的理论基础。
2、利尿剂
当失代偿性右心衰竭导致液体潴留、中心静脉压升高、肝脏淤血、腹水和外周水肿时,可使用利尿剂,以改善症状。
3、氧疗
低氧刺激可引起肺血管收缩、红细胞增多而血液黏稠、肺小动脉重构加速IPAH的进展。伴有低氧血症的IPAH患者应给予氧疗以保持其动脉血氧饱和度持续大于90%。
4、地高辛
地高辛能迅速改善IPAH的心排出量,并可用于降低PAH患者发生房性快速心律失常的心室率。
5、贫血和铁状态
铁缺乏与运动能力下降有关,也可能与高死亡率相关,应对患者进行常规的铁状态监测,如有铁缺乏应继续找寻病因,并补充铁制剂。
6、血管扩张药
(1)钙通道阻滞剂(CCB):急性血管反应试验结果阳性是应用CCB治疗的指征。主要包括硝苯地平、地尔硫卓、氨氯地平,需要在治疗3-4月后重新评估其适用性。
(2)前列环素类似物及受体激动剂:常用的前列环素类似物有:依前列醇、伊洛前列素、贝前列素。另外还有前列环素受体激动剂:噻拉西哌。
(3)一氧化氮(NO):NO吸入是一种仅选择性地扩张肺动脉而不作用于体循环的治疗方法。但是由于NO的作用时间短,加上外源性NO的毒性问题,从而限制了其在临床上的使用。
(4)内皮素受体拮抗剂:常用内皮素受体拮抗剂有:波生坦、安立生坦、马西替坦等。
(5)磷酸二酯酶-5抑制剂:包括西地那非、他达拉非、伐地那非等。
(6)可溶性鸟苷酸环化酶(sGC)激动剂:利奥西呱等。
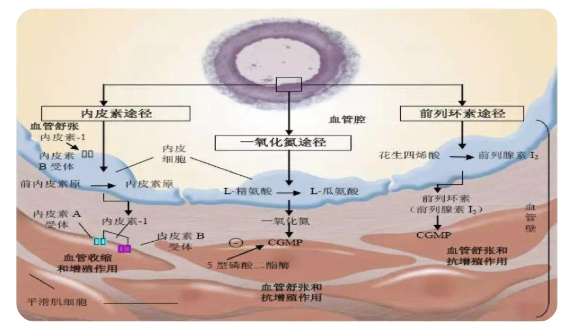
肺动脉高压靶向药物治疗机制
(引自N Engl J Med 2004;351:1425-1436)
肺或心肺移植
经积极内科治疗临床效果不佳的患者可以行肺移植治疗。如同时伴有心脏结构或功能出现不可逆损害者可考虑行心肺联合移植。
健康指导
对IPAH患者进行生活指导,加强相关卫生知识的宣传教育,增强患者战胜疾病的信心,预防肺部感染。
翟振国教授点评
肺动脉高压并不是一个少见的临床疾病,每年的5月5日是世界肺动脉高压日。它应该被更多人认识。肺动脉高压病因涉及多个学科,要求临床医师应当具备多学科的知识与灵活的鉴别诊断思维。
一、重视肺动脉高压的病因筛查
对于每一个肺动脉高压患者,规范的病因筛查流程非常重要,呼吸科医师应该考虑到心血管疾病因素,心脏科医师也不可忘记缺氧与肺部疾病的原因,结缔组织和其他相关疾病相关的肺动脉高压更不应被忽视。对于原因复杂的患者,多学科甄别与共同诊治尤为重要。
临床上,肺动脉压升高的情况并不少见,很多情况下部分医师和患者会认为得了一种「不治之症」。事实上,对于大部分超声检测到肺动脉压升高者,应该首先结合病史与临床表现确定可能的病因筛查方向。
如为老年人或有冠心病、房颤病史的患者,结合超声存在左室收缩或舒张功能减退,或心室流出道梗阻等异常时,往往提示该患者出现左心疾病所致的肺动脉高压。应适当进行强心、利尿治疗,改善心功能后即可取得很好的疗效。若是长期吸烟,有慢性咳嗽、喘憋的患者,则应考虑因低氧和慢性肺病所致的肺动脉高压,给予支气管扩张剂、氧疗等措施,往往会取得奇迹般的效果。还有的患者存在手术、创伤、下肢静脉曲张等病史,可考虑行CT肺动脉造影和肺通气灌注显像;如果有血栓征象,则要考虑慢性血栓栓塞性肺动脉高压(CTEPH),这时不要轻易直接推荐靶向药物或手术治疗,应先给患者3个月充分抗凝治疗后再评估下一步方案。
二、重视肺动脉高压的基础治疗和病因处理
对肺动脉高压患者的治疗不应局限于单纯的靶向药物,而应是一套完整的治疗策略,包括对病情严重程度的评估及对治疗反应的评价等。病因明确的肺动脉高压患者须针对病因治疗;某些患者可能会有几种病因同时存在,这时需要针对多种病因进行治疗;另一方面,基础治疗如吸氧、抗凝、强心、利尿等也不可忽视。
在肺动脉高压患者中,科学合理的抗凝是治疗的重要环节。有研究者在特发性肺动脉高压(IPAH)患者的尸检中发现原位血栓形成的发生率较高,心衰、制动等因素会进一步增加血栓栓塞的风险,这些都是PAH接受口服抗凝药物治疗的理论基础。同时,对于肺栓塞因为抗凝不规范而导致血栓复发、甚至发展为CTEPH的病例屡见不鲜。
病因治疗和基础治疗往往会显著改善患者症状,切忌在没有进行基础治疗时,就盲目地进行靶向药物治疗。其他的一般措施还包括氧疗、利尿等措施的合理使用,以及体力活动和监督下的康复、避免妊娠、绝经后激素治疗、社会心理支持等。
三、重视肺动脉高压的综合管理
肺动脉高压对患者及其家庭的心理、社会、经济等方面具有重大影响,因此欧洲心脏病学会/欧洲呼吸病学会(ESC/ERS)的新版《肺动脉高压诊断和治疗指南》中更强调综合管理的重要性。管理患者的团队应具有处理所有问题的能力和专业知识,根据症状的严重程度与相关方面的专业人员(如精神病科、心理科、福利和社会工作者)保持密切联系。患者支持团体也起着非常重要的作用。
IPAH作为一类容易恶化的临床综合征,较易发生急性失代偿性心功能不全,很难预测患者何时会加重或死亡。临床医师应和患者及家属进行充分的交流,并制定下一步治疗计划。同时加强心理护理和关爱,尽可能让患者保持乐观积极的心态。肺动脉高压患者终末期的治疗更需要频繁进行多学科评估,尽量减少患者的痛苦,选择最合适的药物。此时,心理学、社会学和精神支持等综合管理必不可少。
对肺动脉高压的管理需要不同医学团体、医疗组织及社会机构、患者及家属的通力合作,这一特点反映了肺动脉高压的多学科本质。临床医师有责任针对不同个体的疾病情况做出临床决策,并与患者及照护人员共同协商选择合适方案,更大限度地让患者得到最适宜的治疗和综合管理。
四、新型靶向药物的研发和介入手术技术的进步为肺动脉高压带来更多的希望
20世纪90年代以前,医学界对肺动脉高压确实缺少有效的治疗手段。随着近年来新型药物的研发及临床应用,基因治疗、手术治疗等也不断取得进步,肺动脉高压患者有了更多的治疗选择,患者的平均生存率显著提高。多数肺动脉高压患者有方可治,有路可循,有些患者甚至可以治愈。
1、新型血管活性药物的开发为肺动脉高压患者带来了希望:近年来,开发了许多针对肺动脉高压发病机制的特异性靶向治疗药物,包括内皮素受体拮抗剂、磷酸二酯酶-5抑制剂、前列环素及其类似物、鸟苷酸环化酶激动剂等。这些药物的广泛应用,已经明显地改善了患者的预后,尤其是对PAH和CTEPH。靶向治疗越早患者获益越大。越来越多的试验证据表明,PAH的早期治疗可以明显改善患者的预后,一旦发现患者为PAH,即使处于WHO功能分级Ⅱ级,亦应尽早开始靶向治疗的评估,以延长患者的生存期,提高患者的生活质量。
2、外科手术技术的发展使某些类型的肺动脉高压治愈成为可能:肺动脉血栓内膜剥脱术是治疗CTEPH的一种极为有效的手段,很多患者可通过手术治愈。截至目前,美国圣地亚哥医学中心已经完成了接近4000例手术,手术死亡率已经降至3%以下。国内几家医学中心也已成功开展此项手术,并取得了很好的治疗效果。由心脏外科、呼吸内科、麻醉科、影像科等组成的CTEPH诊断与治疗团队,是手术顺利开展与成功完成的重要保障。肺移植或心肺联合移植手术也已经成为非常成熟的治疗手段,国内肺移植技术不断进步,不仅可以延长IPAH患者的生命,并可极大限度地改善其生活质量;手术成功率可达90%以上,3年和5年生存率分别在70%和60%以上,多数患者术后可恢复正常工作。随着抗排异药物的更新,肺移植患者生存率越来越高。
参考文献
[1] Galiè N, Humbert M, Vachiery JL,et al; ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1;37(1):67-119.
[2] Andrea Frump, Allison Prewitt, Mark P. de Caestecker. BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series). Pulm Circ. 2018 Apr; 8(2): 2045894018765840.
[3] Todd M. Tartavoulle, Aryn C. Karpinski, Andrew Aubin, Benzi M. Kluger, Oliver Distler, Lesley Ann Saketkoo. Multidimensional fatigue in pulmonary hypertension: prevalence, severity and predictors. ERJ Open Res. 2018 Jan; 4(1): 00079-2017.
作者介绍
