
 分享
分享
推荐语
近日,国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布了《第一批罕见病目录》,共收录121种疾病,其中多种为呼吸疾病,为此《呼吸界》将特约专家对此逐一进行分析解读,敬请关注。
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是一种慢性、进行性、纤维化性间质性肺炎。迄今为止,病因不明,故称为特发性。
流行病学与危险因素
IPF是一种罕见疾病,好发于老年人,其患病率和年发病率分别是14~42.7/10万人口和6.8~16.3/10万人口。近来研究发现其患病率呈现上升趋势。我国缺乏相应的流行病学资料,但是临床实践中发现近年来IPF的病例呈明显增多的趋势。
危险因素包括吸烟和环境暴露(如金属粉尘、木尘等),吸烟指数超过20包/年,患IPF的危险性明显增加。还有研究提示了IPF与病毒感染(如 EB病毒)的关系,但是病毒感染在IPF的确切作用不明确。IPF常合并胃食管反流(gastroesophageal reflux,GER),提示胃食管反流致微小吸入可能与IPF发病有关,但是二者之间的因果关系还不十分清楚。家族性IPF病例的报道提示IPF存在一定的遗传易感性,但是还没有特定的遗传异常被证实。
病理改变
IPF的本质是肺脏发生了纤维化改变,这种病理改变的特征是病变呈斑片状分布,主要累及胸膜下外周肺腺泡或小叶。低倍镜下病变呈时相不一,表现纤维化、蜂窝状改变、间质性炎症和正常肺组织并存,致密的纤维斑痕区伴散在的成纤维细胞灶。这种特征性病理改变称为普通型间质性肺炎(usual interstitial pneumonia, UIP)。UIP是IPF的特征性病理改变类型。
发病机制
IPF的病因和发病机制都还不是很清楚。目前认为IPF起源于肺泡上皮反复发生微小损伤后的异常修复。在已知或未知的遗传/环境因素的多重持续损伤下,受损的肺上皮细胞启动「重编程」,导致细胞自噬降低,凋亡增加,上皮再生修复不足,残存细胞发生间充质样转化,呈现促纤维化表型,大量分泌促纤维化因子,形成促纤维化微环境,使成纤维细胞(fibroblasts)活化转变为肌成纤维细胞(myofibroblasts),产生过量的细胞外基质沉积,导致纤维疤痕与蜂窝囊形成,肺结构破坏和功能丧失。
临床表现
多于50岁以后发病,呈隐匿起病,主要表现活动性呼吸困难,渐进性加重,常伴干咳。全身症状不明显,可以有不适、乏力和体重减轻等,但很少发热。75%有吸烟史。
约半数患者可见杵状指,90%的患者可在双肺基底部闻及吸气末细小的Velcro啰音。在疾病晚期可出现明显发绀、肺动脉高压和右心功能不全征象。
辅助检查
1、胸部HRCT
胸部X片诊断IPF的敏感性和特异性差。胸部HRCT可以显示UIP的特征性改变 (图1) ,诊断UIP的准确性大于90%,因此HRCT已成为诊断IPF的必要手段,可以替代外科肺活检。HRCT的影像学诊断分为三级,包括典型UIP表现,可能UIP表现和不符合UIP表现 (见表1) 。
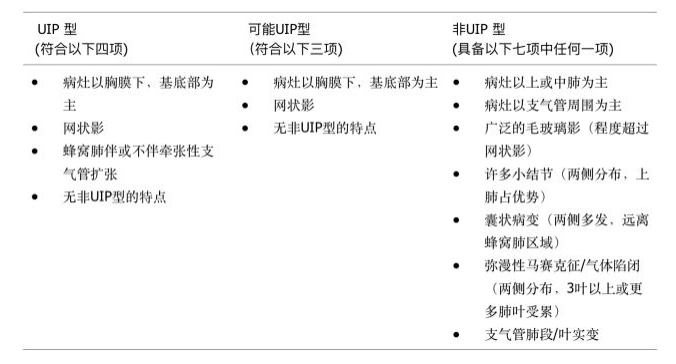
【表1】UIP的HRCT诊断分级标准

图1
2、肺功能
主要表现限制性通气功能障碍、弥散量降低伴低氧血症或I型呼吸衰竭。早期静息肺功能可以正常或接近正常,但运动肺功能表现P(A-a)O2增加和氧分压降低。
3、血液化验
血液KL-6增高,ESR、抗核抗体和类风湿因子可以轻度增高,但没有特异性。结缔组织疾病相关自身抗体检查有助于IPF的鉴别。
4、BALF/TBLB
BALF细胞分析多表现中性粒细胞和/或嗜酸粒细胞增加。BAL或TBLB对于IPF无诊断意义。
5、外科肺活检
对于HRCT呈不典型UIP改变,诊断不清楚,没有手术禁忌证的患者应该考虑外科肺活检。UIP的病理诊断分级标准如表2。
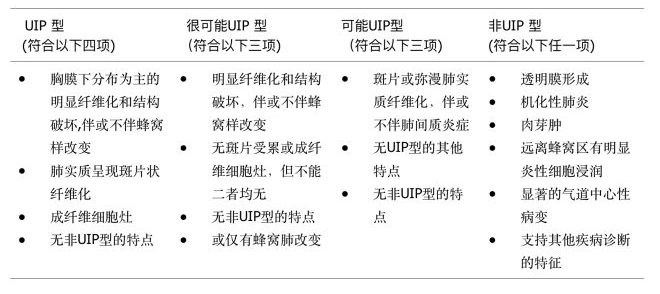
【表2】UIP的病理诊断分级标准
诊断与鉴别诊断
1、IPF诊断遵循如下标准:
① 诊断间质性肺疾病(ILD),但排除了其他原因(如环境、药物和结缔组织疾病等);② HRCT表现为UIP型或 ③ 联合HRCT和外科肺活检病理表现诊断UIP。IPF的临床诊断需要采用排除法和多学科联合会诊,图2描述了IPF的诊断流程。

图2
2、IPF急性加重(acute exacerbation of IPF)
IPF患者出现新的弥漫性肺泡损伤导致急性或显著的呼吸困难恶化即为AE-IPF。诊断标准:①过去或现在诊断IPF;②一月内发生显著的呼吸困难加重;③CT表现为UIP背景下出现新的双侧磨玻璃影伴/不伴实变影;④不能完全由心衰或液体过载解释。
3、鉴别诊断
IPF的诊断需要排除其他原因的ILD。UIP是诊断IPF的金标准,但UIP也可见于慢性过敏性肺炎、石棉沉着病、结缔组织疾病(CTD)等。过敏性肺炎多有环境抗原暴露史(如饲养鸽子、鹦鹉等),BAL细胞分析显示淋巴细胞比例增加。石棉沉着病、硅沉着病或其他职业尘肺多有石棉、二氧化硅或其他粉尘接触史。CTD多有皮疹、关节炎、全身多系统累及和自身抗体阳性。
治疗
IPF不可能治愈,治疗目的是延缓疾病进展,改善生活质量,延长生存。包括抗纤维化药物治疗、非药物治疗、合并症治疗、姑息治疗、疾病的监测、患者教育和自我管理。
1、抗纤维化药物治疗:循证医学证据证明吡非尼酮(pirfenidone)和尼达尼布(Netadenib)治疗可以减慢IPF肺功能下降,为IPF患者带来希望。吡非尼酮是一种多效性的吡啶化合物,具有抗炎、抗纤维化和抗氧化特性。尼达尼布是一种多靶点络氨酸激酶抑制剂,能够抑制血小板衍化生长因子受体(PDGFR)、血管内皮生长因子受体(VEGFR)以及成纤维细胞生长因子受体(FGFR)。两种药物作为抗纤维化药物已开始在临床用于IPF的治疗。N-乙酰半胱氨酸,作为一种祛痰药,高剂量(1800mg/d)具有抗氧化,进而抗纤维化作用,部分IPF患者可能有用。
2、非药物治疗:IPF患者尽可能进行肺康复训练,静息状态下存在明显的低氧血症(PaO2<55mmHg)患者还应该实行长程氧疗,但是一般不推荐有创机械通气治疗IPF所致的呼吸衰竭。
3、肺移植:是目前IPF最有效的治疗方法,合适的患者应该积极推荐肺移植。
4、合并症治疗:积极治疗合并存在的胃-食管反流及其他合并症。
5、IPF急性加重的治疗:由于IPF急性加重病情严重,病死率高,虽然缺乏随机对照研究,临床上仍然推荐相对高剂量激素治疗。氧疗、防控感染、对症支持治疗是IPF急性加重患者的主要治疗手段。一般不推荐有创机械通气治疗IPF所致的呼吸衰竭,但酌情可以使用无创机械通气。
6、对症治疗:减轻患者因咳嗽、呼吸困难、焦虑带来的痛苦,提高生活质量。
7、加强患者教育与自我管理:建议吸烟者戒烟,预防流感和肺炎。
自然病程与预后
IPF诊断后中位生存期为2至3年,但IPF自然病程及结局个体差异较大。大多数患者表现为缓慢逐步可预见的肺功能下降;少数病人在病程中反复出现急性加重;极少数病人呈快速进行性发展。影响IPF患者预后的因素包括:呼吸困难、肺功能下降和HRCT纤维化和蜂窝样改变的程度,六分钟步行试验(6MWT)的结果,尤其是这些参数的动态变化。基线状态下DLco<40%预计值和6MWT时SPO2<88%, 6至12个月内FVC绝对值降低10%以上或DLco绝对值降低15%以上都是预测死亡风险的可靠指标。
参考文献
[1] An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011. 183(6): 788-824
[2] 特发性肺纤维化诊断和治疗中国专家共识,中华结核和呼吸杂志 2016;39(6):427-432
作者介绍
