分享
分享
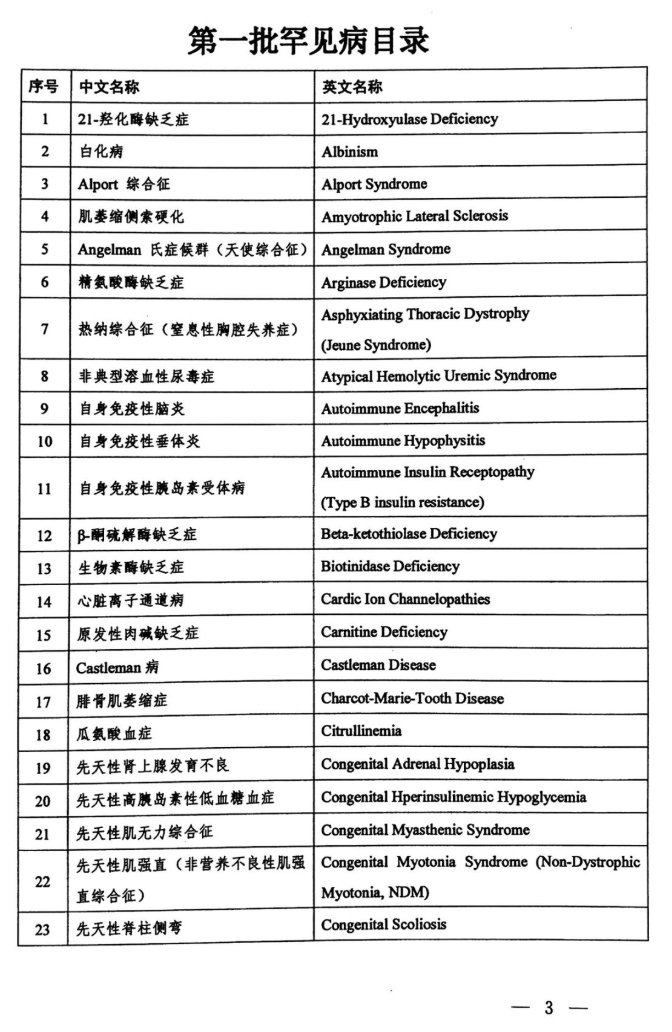
近日,国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布了《第一批罕见病目录》,共收录121种疾病。
根据世界卫生组织的定义,患病人数占总人口0.65‰~1‰的疾病即可被定义为罕见病。目前,全球已知的罕见病超过7000种,我国各类罕见病患者早已超过千万人,由于市场需求小、科研投入少、研发成本高等诸多因素,愿意关注罕见病治疗药物研发的企业并不多,仅有5%的罕见病有治疗方法,大多数患者依然面临着「难以确诊、无药可治、医保未覆盖」的困境,因此,完善相关政策法规、明确罕见病在我国的官方定义等迫在眉睫。
《第一批罕见病目录》将对制定罕见病相关政策产生重要参考价值、有利于推动更多罕见病药物的研发与审批,我们期待这些里程碑式举措尽早惠及更多罕见病患者。
在本次公布的罕见病目录中,有5种疾病与呼吸学科相关:特发性肺动脉高压、特发性肺纤维化、朗格汉斯细胞增生症、淋巴管肌瘤病、肺泡蛋白沉积症,其详细解读且看下文。
特发性肺动脉高压(54)
特发性肺动脉高压(IPAH)是一种不明原因的肺血管阻力增加,从而引起肺动脉压力持续升高,最终导致右心衰竭甚至死亡的疾病。该病发病率较低,但预后极差。西方关于IPAH流行病学资料统计相对较多,欧洲统计显示IPAH的发病率约为1~7.6/100万人。法国数据显示,PAH患者中有39.2%是IPAH患者。我国由阜外医院牵头的全国多中心注册登记研究,2007年至2010年共入组150例IPAH患者。分析结果显示,IPAH在我国多累及青壮年,女性多见,多数患者就诊时心功能分级较差,血流动力学状况不佳。目前国内关于IPAH生存率尚无明确报道,但西方数字显示2007~2009年较2001~2003年IPAH生存率显著提高(HR 1.96;P =0.019)。随着IPAH治疗技术的提高,IPAH预后将有所改善。
(参考文献:Am. J. Respir. Crit. Care Med., 2012, 186(8): 790-6. 中华心血管病杂志,2012,40(8):657-661.)
特发性肺纤维化(55)
特发性肺纤维化(IPF)是一种慢性、进行性、纤维化性间质性肺疾病,病变局限在肺脏,好发于中老年人群,其肺组织学和/或胸部高分辨率CT(HRCT)特征性表现为普通型间质性肺炎(UIP),病因不清。据统计,每年整体人群中的患病率约(2~29)/10万,且呈逐渐增长趋势,估计以每年11%的比例增长。在美国特发性肺纤维化患者大约有100000人,欧盟地区大约有110000人,而且每年欧盟地区新增IPF患者35000人。日本每年整体人群中的IPF患病率约(2.23~10)/10万,实际值远高于这个数目。我国作为一个老龄化严重的国家,目前IPF患病人数也是逐年增加,保守估计至少在50万左右。作为一种慢性间质性肺病,IPF起病隐匿、病情逐渐加重,也可表现为急性加重。IPF诊断后的平均生存期仅2.8年,死亡率高于大多数肿瘤,IPF被称为一种「类肿瘤疾病」。
(参考来源:见评论区)
朗格汉斯细胞增生症(60)
朗格汉斯细胞组织细胞增多症(LCH)是一类罕见的、原因不明的、以组织细胞浸润为特征的疾病,可以累及多个器官系统。多见于婴幼儿或青少年,成人少见。肺LCH可原发于肺脏,也可是全身性系统性病变的一部分,以青年人居多。1997年国际组织细胞协会对LCH的临床分类达成一致意见,将LCH分为单一器官受累、单一系统多部位受累和多系统受累病变型。主要发生于成年人的以肺部为唯一受累器官的肺LCH,与全身系统受累型肺部受累的LCH的病程及预后不同,可能代表两种不同的疾病过程。目前认为,肺LCH是一种少见的与吸烟相关的弥漫性肺实质疾病,肺活检资料表明肺LCH约占肺简质病的5%,该病与吸烟密切相关,统计数据显示约90%以上的病例有吸烟史朗格汉斯细胞组织细胞增多症,分为单个器官受累、多器官受累和多系统受累。PLCH有自限倾向,75%的患者在戒烟后6~24个月病情稳定或好转。也有部分患者预后较差,出现其它脏器受累,导致不良转归。该病症状不典型,成人易被漏诊误诊。目前国内诊断例数极低,多为个案报道。
(参考文献:中华结核和呼吸杂志,2015,(5):393-395. )
淋巴管肌瘤病(64)
淋巴管肌瘤病(LAM)是一种比较罕见的疾病,以育龄期女性为主,平均发病年龄30~40岁。LAM主要累及肺脏,典型表现为弥漫性囊性改变,严重影响患者肺功能,而目前尚缺乏有效的治疗方法。该病流行学统计数字多来源于西方及日本,发病率约为1~7.5/百万人,10年生存率约为76~91%。国内报道多为个案病例报道。
(参考文献:Adv. Exp. Med. Biol., 1031, 419-442.)
肺泡蛋白沉积症(100)
肺泡蛋白沉积症(PAP)是一种以肺泡腔内积聚过多过碘酸雪夫染色阳性的脂蛋白物质为特征的临床罕见疾病。临床上将PAP分为3类:先天性、特发性及继发性PAP,其中90%以上是特发性PAP(IPAP),IPAP的发病机制并不完全清楚,目前认为是一种自身免疫性PAP。该病临床表现差异明显,患者可无症状,也可出现呼吸衰竭而危及生命,全肺灌洗是目前治疗该病的主要手段。该病多于中年起病,国际多中心研究统计结果所示,发病率约6.87/百万人。该病预后差异较大,部分患者可自行缓解,也有患者可因急性呼吸衰竭、感染致死。
(参考文献:中华结核和呼吸杂志,2004,(12):824-828. Adv. Exp. Med. Biol., 1031, 419-442.)
下方为文件全文: