
 分享
分享
摘要
背景和目的:近年来,免疫检查点抑制剂(Immune checkpoint inhibitors, ICIs)成为治疗晚期非小细胞肺癌(non-small cell lung cancer, NSCLC)患者的重要策略。肿瘤新抗原作为重要的生物标志物和潜在的免疫治疗靶点,在NSCLC患者的治疗和预后等方面发挥着重要作用。本研究旨在评估非小细胞肺癌的手术标本中肿瘤体细胞突变与潜在新抗原之间的相关性。
方法:本研究为前瞻性研究,收集了2019年6月至2019年9月在北京协和医院胸外科接受肿瘤手术切除治疗的非小细胞肺癌患者。对入组患者肿瘤组织和相对应的正常组织进行全外显子测序(Whole exome sequencing, WES),通过生信软件对候选新抗原进行预测,分析新抗原表达的基因突变特征。
结果:与所有突变基因相比,新抗原相关基因突变种类较少;与低肿瘤新抗原负荷(tumor neoantigen burden, TNB)相关基因相比,高TNB相关基因突变种类更多,同时突变频率更高;在突变类型中,候选新抗原数和错义突变、移码插入/缺失、剪接位点变异和无义突变呈正相关,而在多元线性回归分析中,只有错义突变和新抗原呈正相关有统计学差异;此外,碱基颠换(A>C/C>A、T>G/G>T、C>G/G>C)和新抗原表达量呈正相关,而碱基转换(A>G/G>A和C>T/T>C)和新抗原表达量呈负相关。
结论:在NSCLC标本中,候选新抗原的表达量与基因突变频率、突变类型和碱基替换类型相关。
关键词:非小细胞肺癌;全外显子测序;新抗原;肿瘤突变负荷;基因突变特征

前言
尽管肺癌的治疗手段在过去20年里取得了进展,但其仍然是全球癌症相关死亡的主要原因。近年来,针对细胞毒性T淋巴细胞相关蛋白4(cytotoxic T-lymphocyte-associated protein 4, CTLA-4)、程序性死亡受体-1(programmed cell death 1, PD-1)和程序性死亡受体-配体1(programmed cell death-Ligand 1, PD-L1)的免疫检查点抑制剂(Immune Checkpoint Inhibitors, ICIs)在晚期非小细胞肺癌(non-small cell lung cancer, NSCLC)中显示出持久的临床获益,彻底改变了晚期NSCLC患者的治疗标准选择。尽管ICIs与化疗相比在一线和二线方案中显示出生存优势,但在未经选择的患者中客观反应率(Objective Response Rate, ORR)仅为20%左右[1-11]。因此,筛选出可能从ICI治疗中获益的人群至关重要。
目前PD-L1表达是唯一被批准用于ICIs治疗的生物标志物,但PD-L1低表达的患者也可获得治疗反应,表明其还不足以完全筛选出免疫治疗可能获益人群[12]。肿瘤免疫其它潜在的生物标志物包括MHC表达、淋巴细胞计数、肿瘤T细胞标志物表达、肿瘤突变负荷(tumor mutation burden, TMB)和肿瘤新抗原等[13]。其中,突变衍生的新抗原作为一种潜在的免疫治疗的生物标志物引起了广泛的关注。新抗原是肿瘤特异性的突变肽,可由主要组织相容性复合物(major histocompatibility complex, MHC)分子递逞[14, 15],并被T细胞识别,从而介导对肿瘤细胞的免疫反应,使宿主免疫系统识别和破坏肿瘤细胞。
近年来,基因组学和生物信息学的技术进步为有效地从肿瘤的体细胞突变谱中选择最强的免疫原性新抗原奠定了基础。然而,目前对于非小细胞肺癌新抗原相关的基因突变特征的报道相对较少,而这一特征在指导临床筛选免疫获益人群方面也至关重要。因此,本文旨在评估非小细胞肺癌的手术标本中肿瘤体细胞突变与潜在新抗原之间的相关性。
患者和方法
患者
收集2019年6月至2019年9月在北京协和医院胸外科接受手术切除治疗的非小细胞肺癌患者。入组标准如下:①组织病理学确诊为非小细胞肺癌;②有完整的临床病理资料;③有足够的肿瘤组织和正常组织接受全外显子测序;④患者已签署知情同意书。统计入选患者的性别、年龄、吸烟史、组织病理学类型、TNM分期和临床分期。该研究已经通过了北京协和医院的伦理审核。
新抗原预测
通过对非小细胞肺癌肿瘤组织和相对应正常组织的全外显子测序结果进行分析,结合主要组织相容复合物(major histocompatibility complex, MHC )I类分子亲和力的预测和基因表达量,使用生信软件(Neopipe Software)预测候选新抗原(见图1)。由于没有RNA数据,基因表达量参考TCGA数据库非小细胞肺癌的相关数据(见图2)。对于不同病理类型,肺腺癌和肺鳞癌分别参考574个肺腺癌数据和548个肺鳞癌数据的均值作为转录本定量和基因定量的结果;其它病理类型参考所有肺癌患者的数据均值作为转录本定量和基因定量的结果。
为了便于体外合成,我们将新抗原的长度设计为25-mer肽。其中可以与MHC结合的8-11-mer肽被定义为新抗原表位。鉴于新抗原表位和MHC亲和力的差异性,仅基于新抗原表位的数量而预测免疫原性的准确性会很低。因此,本文主要分析新抗原相关的基因突变特征。

图1:新抗原预测流程图

图2:基因扰动图
统计学分析
数据采用SPSS 21.0(IBM Corp., Armonk, NY, USA)统计软件进行统计学分析。统计结果以中位数、频数、频率和构成比表示;采用非参数检验分析与候选新抗原表达相关的临床病理特征;采用Spearman检验分析候选新抗原的基因突变特征;采用多元线性回归分析多种与候选新抗原相关的基因突变类型;采用Rstudio软件绘制热图及进行相关聚类分析;双侧检验,P值<0.05认为有统计学差异。
结果
临床病理特征以及候选新抗原的预测
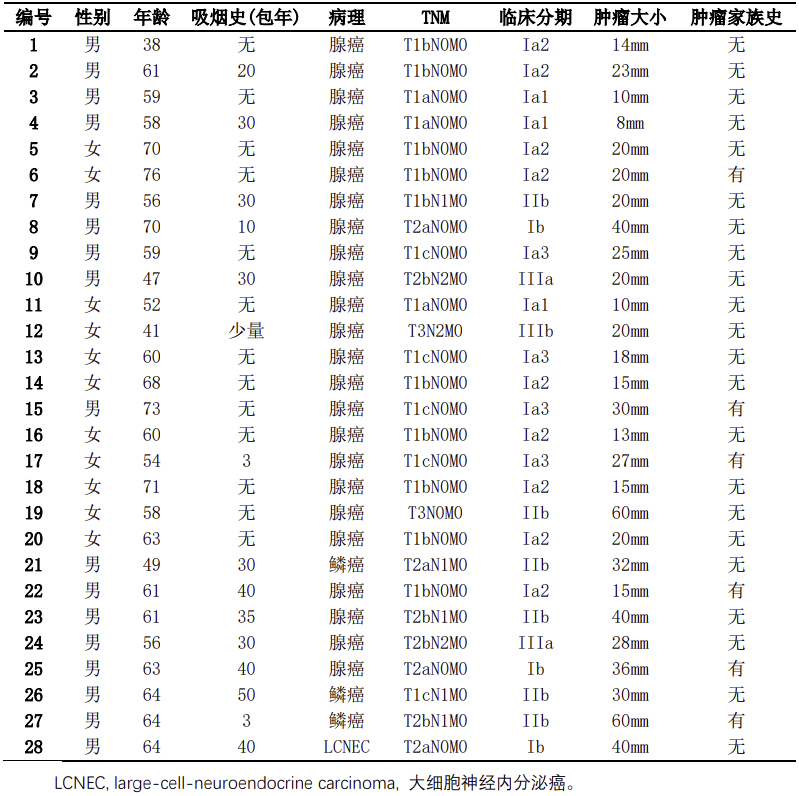
2019年6月至2019年9月共收集接受手术切除治疗的非小细胞肺癌患者34例。排除6例组织标本量不足以及临床资料不全的患者后,最终28例患者纳入研究。28例入组患者中,中位年龄60.5岁(38-76岁);男性患者17例(60.7%);有吸烟史患者15例(53.6%);病理类型:腺癌24例(85.7%),鳞癌3例(10.7%),大细胞神经内分泌癌1例(3.6%);肿瘤分期:I期19例(67.9%),II期6例(21.4%),III期3例(10.7%);有肿瘤家族史6例(21.4%)(见表1)。
表1:入组患者临床病理信息
通过对28例非小细胞肺癌标本进行WES测序,共鉴定出5017个非同义突变,其中包括4037个错义突变,419个移码插入/缺失、313个框内插入/缺失、229个无义突变,10个非终止突变和9个剪接位点变异。
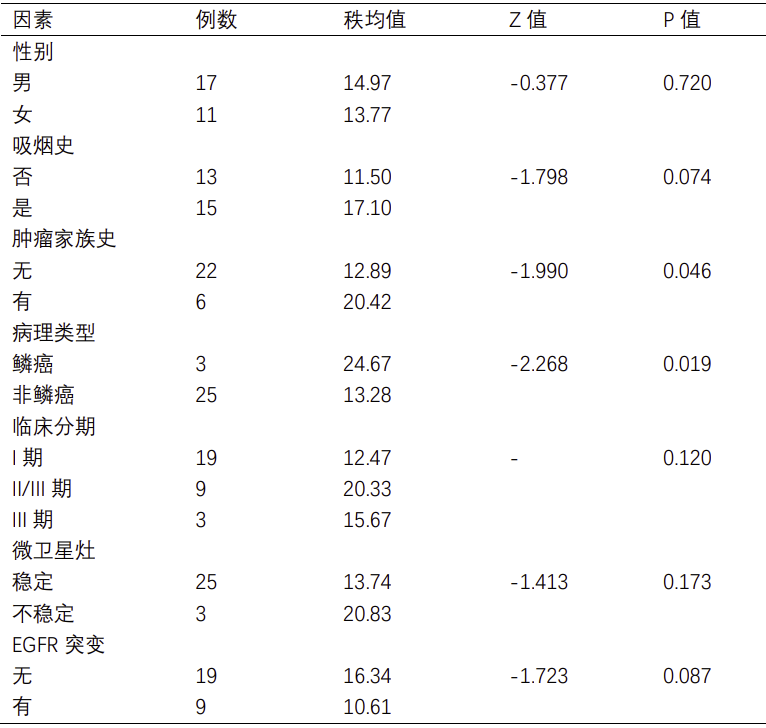
共鉴定出7452个单核苷酸变异(Single nucleotide variation, SNV),包括A>T/T>A(n = 539)、A>C/C>A (n = 966)、A>G/G>A(n = 2006)、T>C/C>T(n = 1990)、T>G/G>T(n = 1025)和C>G/G>C (n = 926)。通过生信软件(Neopipe software)对肿瘤组织进行新抗原的预测,在28例肿瘤组织中,共预测出候选新抗原2942个(中位数为78,范围为28-510),候选新抗原表位(即8-11mer氨基酸的肽段)7912个(中位数为200,范围为48-1300)(见图3)。通过统计学分析,发现肿瘤最长径和候选新抗原表达量呈正相关相关(Spearman相关性分析,相关系数=0.575,P值=0.001);此外,在有肿瘤家族史(秩均值分别为20.42和12.89,P值=0.046)和病理类型为鳞癌(秩均值分别为26.47和13.28,P值=0.019)的患者中,候选新抗原的表达量更高(见表S1)。
28例患者中,在目前常见的肺癌驱动基因中,EGFR驱动基因突变患者9例,其中包括6例21L858R,2例19del和1例20ins,非参数检验分析结果表明候选新抗原负荷和EGFR突变情况无关(P值 = 0.087)(见表S1);EML4-ALK融合患者1例(85个候选新抗原);ROS1融合患者1例(36个候选新抗原);KRAS突变患者3例,包括1例KRAS G12D突变(87个候选新抗原),1例KRAS G12D/CDKN2A D108H共突变(28个候选新抗原)和1例KRAS G12V/TP53 K132E/STK11 N181Y共突变(85个候选新抗原)。
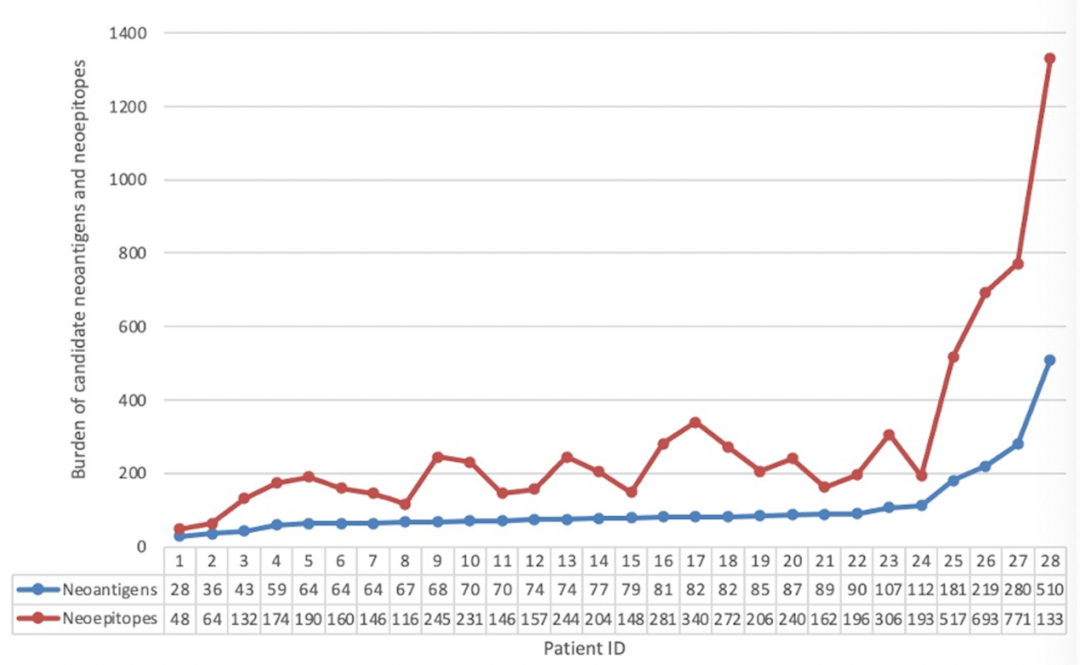
图3:入组患者新抗原表位和新抗原分布图
表S1:预测新抗原数目的非参数检验分析
候选新抗原的基因突变特征
通过统计分析28例患者所有基因突变情况和候选新抗原相关基因的突变情况,我们发现在所有突变基因中,最常见的前10位突变基因类型包括MUC17 (57%)、AHNAK (54%)、ANKRD36C (54%)、HERC2 (50%)、ZNF208 (50%)、ZNF729 (50%)、AHNAK2 (43%)、MUC16 (43%)、CDC27 (39%)和MUC12 (39%)(见图4A);候选新抗原相关的基因突变类型中最常见的前10位突变基因包括CDC27 (29%)、HERC2 (25%)、MUC16 (21%)、ANKRD36C (21%)、BCLAF1 (18%)、GPR32 (18%)、MUC12 (18%)、MUC17 (18%)、PBMX (18%)和TTN (18%)(见图4B)。在所有突变基因中,突变频率更高,突变的种类更多,包括错义突变、无义突变、框内缺失、移码缺失、框内插入、移码插入和混合突变;但常见的候选新抗原相关的基因突变种类只包括其中的几种,包括错义突变、无义突变、框内插入、移码插入和混合突变。
与新抗原负荷相关的基因突变特征
我们将肿瘤新抗原负荷 (TNB) 定义为一份肿瘤样本中,每 Mb 碱基变异中能合成的新抗原总数。根据TNB表达情况,将患者分为高TNB组(>中位数,n=14)和低TNB组(≤中位数,n=14),对两组患者的基因突变特征进行分析。结果表明,在高TNB组患者中,最常见的前10位突变基因包括MUC17 (17%)、AHNAK (57%)、ANKRD36C (57%)、HERC2 (57%)、ZNF729 (57%)、AHNAK2 (50%)、CDC27 (50%)、MUC12 (50%)、TTN (50%)和CACNA1A (43%)(见图4C);在低TNB组患者中,最常见的前10位突变基因包括MUC16 (57%)、ZNF208 (57%)、AHNAK (50%)、ANKRD36C (50%)、EGFR (43%)、FLG (43%)、HERC2 (43%)、MUC17 (43%)、ZNF729 (43%)和ZNF91 (43%)(见图4D)。在高TNB组患者中,基因突变频率更高,突变的种类更多,包括错义突变、无义突变、框内缺失、移码缺失、框内插入、移码插入和混合突变,其中混合突变、插入突变和缺失突变的频率明显高于低TNB组患者;在低TNB组患者中,基因突变种类包括错义突变、无义突变、框内缺失、移码缺失、框内插入和混合突变。
因此,与所有突变基因相比,新抗原相关基因的突变种类更少,表明肿瘤新抗原负荷与基因突变的表达量和种类均相关;与低TNB组患者相比,高TNB组患者的基因突变种类更多,提示某些基因突变类型可触发更高丰度的新抗原负荷。
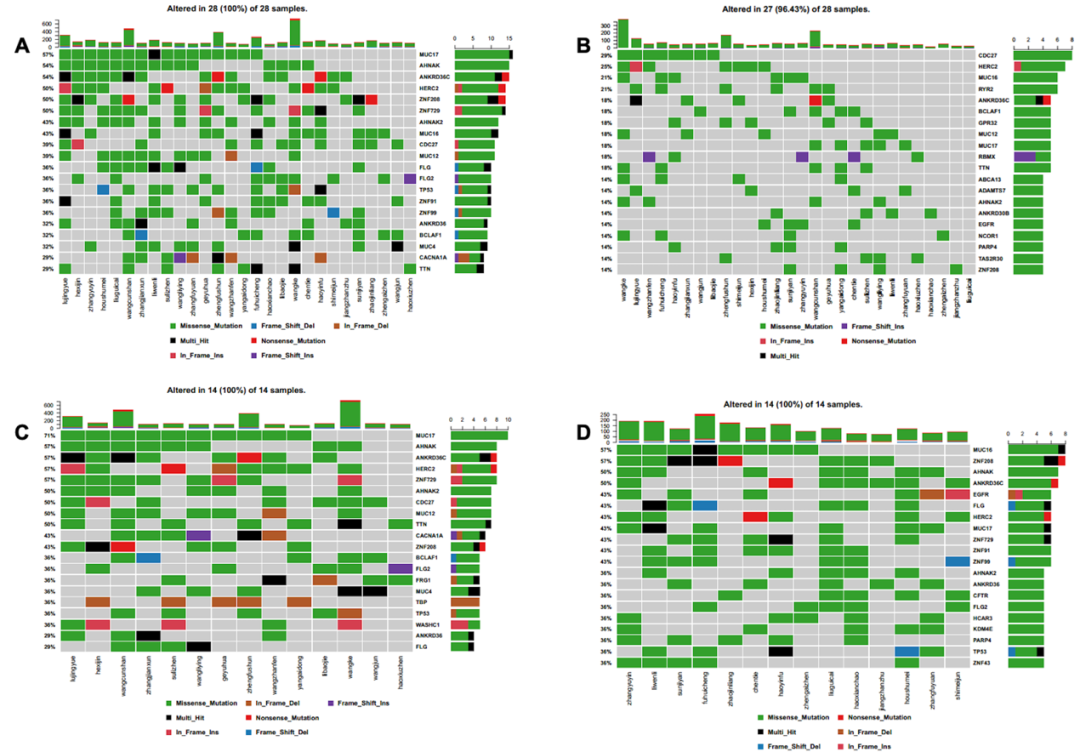
图4:基因突变热图 (A)所有突变基因;(B)新抗原相关的突变基因;(C)高新抗原突变负荷组基因突变;(D)低新抗原突变负荷组基因突变
不同基因突变类型对候选新抗原的影响
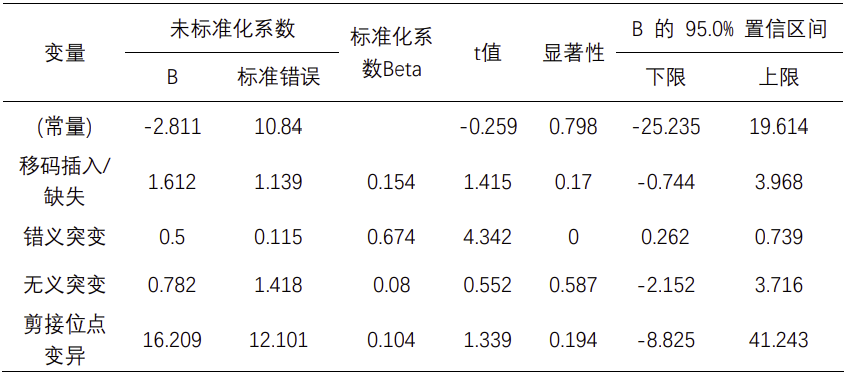
采用spearman检验统计分析新抗原和基因突变类型之间的相关性;结果表明,非同义突变数目(相关系数=0.641,P值<0.001)和候选新抗原表达量呈正相关;随后我们对非同义突变类型进行了注释分析,进一步发现候选新抗原表达量和错义突变(相关系数=0.603,P值<0.001)、移码插入/缺失(相关系数=0.755,P值<0.001)、无义突变(相关系数=0.501,P值 = 0.007)和剪接位点变异(相关系数=0.546,P值 = 0.003)呈正相关(见表2)。接下来,我们将这四种突变类型纳入多元线性回归分析,结果表明候选新抗原表达量仅和错义突变呈正相关有统计学意义(beta = 0.674,P值<0.001)(见表3),这可能与错义突变频数较高相关,而其他突变类型虽然可能触发更高丰度的新抗原负荷,但突变频数较低。同样,我们对碱基替换类型和候选新抗原表达量也进行了spearman相关性分析,结果表明碱基颠换类型,包括A>C/C>A突变频率(相关系数=0.641,P值<0.001)、T>G/G>T突变频率(相关系数=0.388,P值=0.041)以及C>G/G>C突变频率(相关系数=0.418,P值=0.027)和候选新抗原表达量呈正相关,而碱基转换类型,包括A>G/G>A突变频率(相关系数=-0.690,P值<0.001)和T>C/C>T突变频率(相关系数=-0.535,P值=0.003)则与候选新抗原表达量呈负相关(见表2)。
因此,候选新抗原表达量和肿瘤突变频率、肿瘤突变类型均相关,同时和碱基颠换呈正相关,和碱基转换呈负相关。
表2:候选新抗原相关的Spearman相关性分析
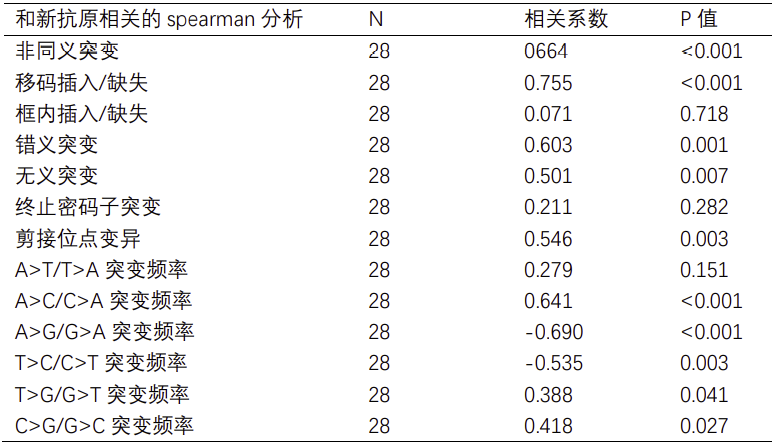
表3:候选新抗原相关对多元线性回归分析
多个候选新抗原相关的基因分析
在与候选新抗原相关的基因改变中,共包括1922个基因,每个基因改变所导致的候选新抗原数目为1-28个不等。我们对生成候选新抗原数≥7的21个基因进行了聚类分析(见图5),结果没有发现候选新抗原表达量和这些基因表达之间存在明显相关性。
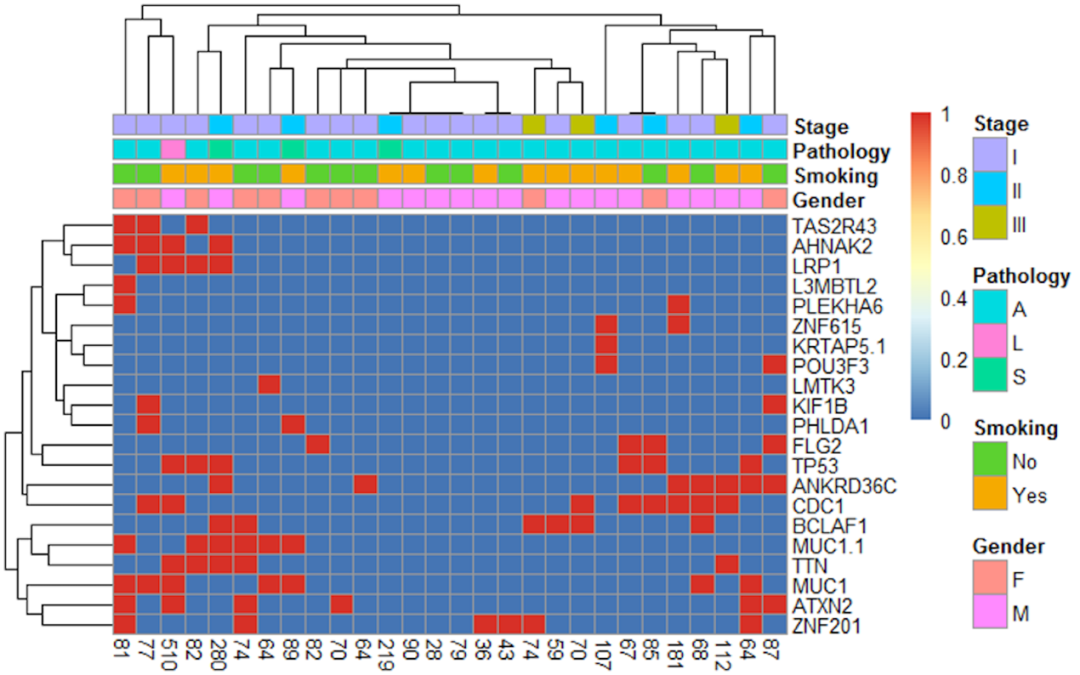
图5:28例患者导致不同候选新抗原个数的基因热图统计
讨论
基于对高TMB NSCLC的ICIs治疗疗效的观察,研究发现TMB是潜在的预测ICIs治疗NSCLC疗效的生物标志物[16]。此外,临床前和临床研究表明,肿瘤特异性错义突变可能产生单个新抗原,介导对免疫检查点抑制剂的应答[14, 17]。故可推测高肿瘤突变负荷会导致更多新抗原的产生,从而增加肿瘤的免疫原性以及对ICIs治疗的反应[18]。这和本研究发现的非同义突变与新抗原表达量相关这一结果相一致。我们通过进一步的分类、注释和分析发现新抗原表达量和错义突变(最常见的突变类型)以及突变频率较少的突变类型如移码插入/缺失、无义突变和剪接位点变异呈正相关。有证据表明[19, 20],与非同义单核苷酸变异相比,移码插入或缺失虽然罕见,但却是一种高度免疫原性的突变类型,它可以通过触发更高丰度的新抗原负荷以及更高的MHC分子亲和力产生更强的免疫原性。
多项研究表明,肿瘤特异性剪接也是新抗原的重要来源[21-23],尽管剪接的表达水平相对较低,但从选择性剪接事件中获得的新抗原比单核苷酸变异更频繁[24];我们发现尽管无义突变突变频率较低,但也和潜在新抗原表达量相关,提示其可能可以触发更高丰度的新抗原负荷。据我们所知,目前尚无对无义突变和新抗原负荷之间关系的报道,还需进一步研究证实。
我们还发现新抗原负荷和碱基颠换呈正相关,和碱基转换呈负相关。既往研究结果表明[25],在接受pembrolizumab治疗的NSCLC患者中,持久临床获益(durable clinical benefit, DCB)的患者含有更多的C>A颠换,以及更少的C>T转换(Mann-Whitney检验, P = 0.01)。这也从侧面印证了我们的研究结果。
之前有研究报道利用TSNAD软件对来自国际癌症基因组协会(International Cancer Genome Consortium, ICGC)数据库的9155个样本的癌症体细胞突变进行了潜在新抗原的预测。发现最常见的潜在新抗原是由KRAS、PIK3CA和TP53基因所编码,在前10位潜在新抗原中,有6种与KRAS基因相关的潜在新抗原,包括两种不同的突变类型:G12D和G12V[26]。此外,一项研究对KRAS突变型肺腺癌进行了基因组、转录组和蛋白质组数据的综合分析[27],确定了三个具有生物学差异的KRAS突变亚组,包括STK11/LKB1 (KL亚型)、TP53 (KP亚型)和CDKN2A/B (KC亚型)。在免疫系统参与方面,KP亚型表现为强烈的炎症反应,体现在包括PD-L1在内的多种共刺激和共抑制因子表达增强。相比之下,KL亚型的肺腺癌免疫标志物的表达量更少。尽管暴露于吸烟的环境相似,与KL亚型肺腺癌相比,KP亚型肺腺癌表现出更高的突变率,这一发现可能有助于解释这两个KRAS突变亚型在免疫原性方面的差异[27]。而我们的研究中,在所有28例患者中仅有3例患者出现了KRAS突变,包括1例KRAS G12V突变(KP和KL混合型,候选新抗原85个),1例KRAS G12D突变(KC亚型,候选新抗原28个)和1例KRAS G12D突变(未合并任何上述提到的突变,候选新抗原87个)。其中只有KC亚型患者的候选新抗原表大量明显低于中位数(78个),另外2例均稍高于中位数。在KRAS突变和新抗原相关性方面,我们并未发现和之前文献报道相一致的结果,这可能与我们的样本量较少相关(KRAS突变患者仅有3例,未达到统计学意义)。
我们的研究也有不足之处,首先,新抗原预测过程中无RNA相关数据,只能以TCGA数据库肺癌基因的表达量作为参考;其次,样本量较少,还需扩大样本量进一步验证研究结果。
结论
我们发现候选新抗原表达量和基因突变频率、突变类型均相关。在和候选新抗原表达量相关的突变类型中,错义突变的频率最高;而移码插入/缺失、剪接位点变异、无义突变的频率较低,提示这些突变类型可能可以触发更高丰度的新抗原负荷。然而,在多元线性回归分析中,只有错义突变和潜在新抗原表达量呈正相关。新抗原表达量和碱基颠换呈正相关,和碱基转换呈负相关。
参考文献
1.Spigel DR, Reckamp KL, Rizvi NA, et al. A phase III study (CheckMate 017) of nivolumab (NIVO; anti-programmed death-1 [PD-1]) vs docetaxel (DOC) in previously treated advanced or metastatic squamous (SQ) cell non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2015;33.
2.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. New Engl J Med. 2015;373:1627-1639.
3.Horn L, Spigel DR, Vokes EE, et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017;35:3924-3933.
4.Wu YL, Lu S, Cheng Y, et al. Nivolumab Versus Docetaxel in a Predominantly Chinese Patient Population With Previously Treated Advanced NSCLC: CheckMate 078 Randomized Phase III Clinical Trial. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2019.
5.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (London, England). 2016;387:1540-1550.
6.Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378:2078-2092.
7.Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379:2040-2051.
8.Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England). 2017;389:255-265.
9.Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med. 2018;378:2288-2301.
10.Antonia SJ, Villegas A, Daniel D, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. New Engl J Med. 2018;379:2342-2350.
11.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823-1833.
12.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature Reviews Cancer. 2016;16:275-287.
13.Penault-Llorca F, Radosevic-Robin N. Tumor mutational burden in non-small cell lung cancer-the pathologist's point of view. Translational lung cancer research. 2018;7:716-721.
14.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74.
15.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413-3421.
16.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-+.
17.Gubin MM, Zhang XL, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577-+.
18.McGranahan N, Furness AJS, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463-1469.
19.Turajlic S, Litchfield K, Xu H, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncology. 2017;18:1009-1021.
20.Linnebacher M, Gebert J, Rudy W, et al. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer. 2001;93:6-11.
21.Hoyos LE, Abdel-Wahab O. Cancer-Specific Splicing Changes and the Potential for Splicing-Derived Neoantigens. Cancer Cell. 2018;34:181-183.
22.Jayasinghe RG, Cao S, Gao QS, et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018;23:270-+.
23.Park J, Chung Y-J. Identification of neoantigens derived from alternative splicing and RNA modification. Genomics & informatics. 2019;17:e23-e23.
24.Kahles A, Ong CS, Zhong Y, Ratsch G. SplAdder: identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics. 2016;32:1840-1847.
25.Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-128.
26.Zhou Z, Lyu XZ, Wu JC, et al. TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection. Roy Soc Open Sci. 2017;4.
27.Skoulidis F, Byers LA, Diao LX, et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discovery. 2015;5:860-877.
原文链接
https://doi.org/10.3389/fimmu.2021.749461
第一作者

梁红格
北京协和医院呼吸与危重症医学科2017级博士研究生,师从王孟昭教授,主要研究方向为肺部肿瘤。已发表多篇核心期刊和SCI论文。
通讯作者

王孟昭
主任医师,教授,博士研究生导师。中国医学科学院北京协和医院呼吸与危重症医学科主任。多年来致力于呼吸道疾病诊治及科研工作。