
 分享
分享
病例介绍
现病史
20岁女性,反复干咳伴活动后气短1年,1周青霉素治疗后无效,伴右胫骨前皮下质硬无痛结节。
胸部CT:双肺多发结节、团块及实变影,两肺门及纵隔多发肿大淋巴结(图1)。
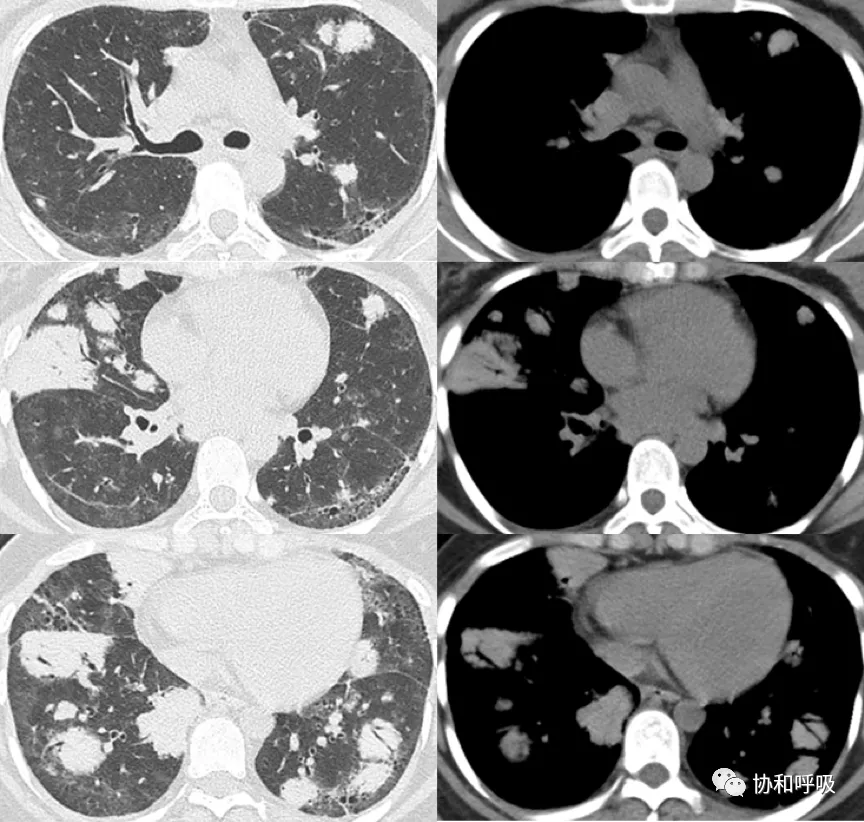
【图1】肺CT提示双肺多发团块
肺穿病理:
肺组织间胶原增生、致密、层状排列,胶原间见透明裂隙,可见少量淋巴细胞、浆细胞浸润,不除外透明变性肉芽肿(PHG)(图2)。
PHG是累及呼吸系统的罕见病变,其病因不明, 有报道称发病年龄约22~77岁,无性别差异,多表现为咳嗽、胸痛、呼吸困难或咯血等,但约有25% 的患者并无症状。近70%患者肺内为多发结节,部分有钙化或空洞,多不累及胸膜,PET/CT可为高代谢病变,也可累及肺外,如皮肤、胸膜、心包、肝等 [1]。病理由致密的以涡轮状或席纹状的方式排列的胶原束构成,期间散在少量炎性细胞。需与结节病、结核、肿瘤等鉴别。
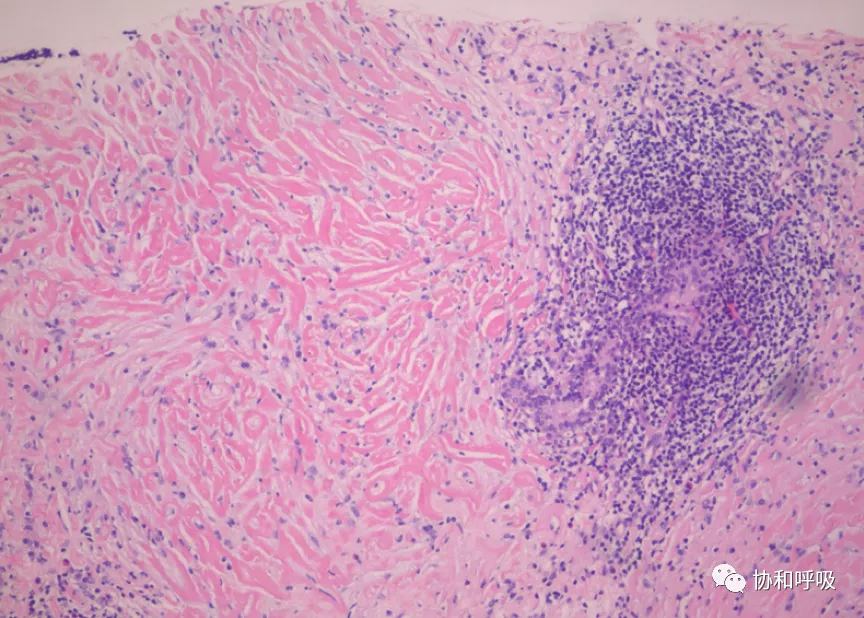
【图2】肺穿刺病理提示大量透明胶原沉积,伴随小血管及淋巴细胞浸润 (HE, ×40).
既往史
诊断特发性血小板减少性紫癜4年,予泼尼松50mg qd口服1月后于1年半后逐渐减停,监测PLT波动于 40-60×10^9/L。
体格检查
不吸氧状态下SpO2 98%;双肺呼吸音清,心腹查体无殊;右胫骨前皮肤陈旧色素沉着。
辅助检查
ESR>140mm/h,hsCRP 150.1mg/L;IgG 27.59g/L;ANA(+)H1:160,Coombs(+);骨髓涂片+活检:符合血小板减少症;肿瘤标记物及气管镜下肺泡灌洗液及毛刷细菌、真菌、结核筛查、肿瘤细胞均阴性;腰穿脑脊液压力200mmH20,常规、生化、细胞学、病原(-)。眼底:光学相干断层成像(OCT)示双眼视乳头轻度水肿,视野检查(VF)示右颞下周边视野缺损,右侧视觉诱发电位(VEP)提示视交叉前损害,考虑视乳头水肿可解释VEP异常和视野缺损。
治疗反应
免疫科会诊:结缔组织病伴自身免疫性血小板减低。
考虑到肺透明变性肉芽肿可伴发结缔组织病,予泼尼松50mg qd、他克莫司1mg bid治疗,后根据他克莫司血药浓度调整剂量;患者半年内逐渐将激素规律减量至5mg qd,但复查肺CT肺内病变无明显变化。
部分PHG患者未经治疗疾病可持续进展,其诊治经验仅来自于个例或少数的病例总结文献,提示部分可伴发自身免疫性疾病,一般来说激素及免疫抑制剂治疗有效[2]。但PHG诊断依赖于病理,尤其是较大块的病理标本,穿刺病理所得到诊断有时是存疑的,并需临床除外感染、淋巴增殖性疾病等其他诊断。
患者最可能的诊断是(单选)
- 原发性肺淀粉样变
- 肺淋巴瘤
- 结缔组织病肺受累
- Castleman病
- 透明变性肉芽肿
进一步检查
免疫固定电泳、血轻链、尿免疫固定电泳及24小时尿轻链均阴性,血清β2微球蛋白 1.620mg/L,IL-6 7.3 pg/mL,HHV-8阴性。
腹部超声:腋下、腹股沟淋巴结超声可见淋巴结。
行胸腔镜左肺活检
肺组织细菌、真菌、结核、奴卡、放线培养均阴性。
病理:肺组织中可见粉染物形成结节,考虑为淀粉样物质沉积。CD3(-),CD20(滤泡),CD138(+),Ki-67(index 1%),Kappa(+),Lambda(+),CD38(+),IgG(-),IgG4(-),刚果红(+),醇化刚果红(+),高锰酸钾化刚果红(+)。
经补取病理切片,可见沿支气管血管及肺膜下分布的增生淋巴组织,淋巴滤泡形成,周边可见较多浆细胞,不除外Castleman病(图2)。
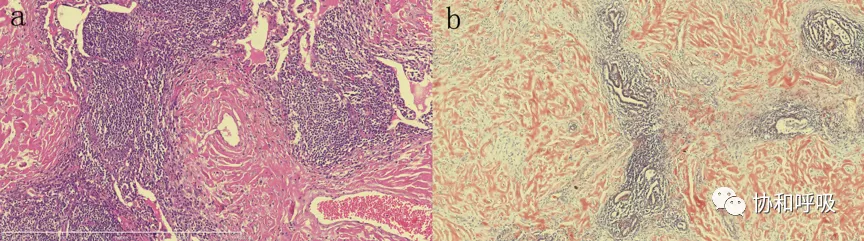
【图3】胸腔镜肺活检 a.肺组织中可见粉染物形成结节,并可见沿支气管血管及肺膜下分布的增生淋巴组织,淋巴滤泡形成,周边可见较多浆细胞(HE, ×200) b. 醇化刚果红(+),考虑为淀粉样物质沉积(醇化刚果红, ×200).
最终诊断
多中心型Castleman病
肺部受累
继发性淀粉样变
血小板减少性紫癜
治疗与转归
血液内科予多程BCD(硼替佐米、环磷酰胺、地塞米松)方案治疗后,PLT水平恢复正常,复查IL-6 2.2 pg/mL,ESR 11mm/h,hsCRP 1.76mg/L,IgG 10.84g/L,但肺内病变无明显变化。
讨论
Castleman病
Castleman病(CD)是病因不清、表现为淋巴结增生的良性疾病,其中浆细胞型和混合细胞型可有不同系统受累表现,如发热、消瘦、贫血、脾大、外周神经病变、高球蛋白血症、急性期炎症性蛋白升高等。
诊断标准
2017年提出了特发的、HHV-8阴性的多中心CD(iMCD)的诊断标准,包括病理确诊的多发淋巴结病变,≥2临床或实验室改变,并除外可模拟iMCD的其他疾病 [3],并定义了「TAFRO综合征」,即伴发血小板减低、水肿或腹水、骨髓纤维化、肾功能不全、器官肿大这一特殊类型的iMCD。
本例小结
CD诊断不易,而CD又可继发如淀粉样变等其他改变,就如本例患者这样。
PHG是一除外性诊断,其诊断需首先需除外感染、肿瘤等其他病因,只有当本例患者得到胸腔镜大块组织标本才得以对增生淋巴组织进行细致观察,并完成免疫组化后,才揭示其粉染物结节本质为淀粉样物质沉积。
多数CD继发的淀粉样变位于肾脏,也可见于其他器官,认为病因为CD引起的慢性炎症,来源于CD生发中心的IL-6可促进淀粉样物质前体的生成,而即使采用积极的CD全身系统性治疗,或尝试放疗、激素、IL-6单抗等,已形成的淀粉样病变仍难以逆转。
本例患者应用BCD化疗后评估CD活动性的炎症指标、IL-6、IgG水平明显下降,临床治疗有效,可避免CD继续进展继发新的肺内淀粉样变的生成。
参考文献
[1] Lhote R, Haroche J, Duron L, et al. Pulmonary hyalinizing granuloma: amulticenter study of 5 new cases and review of the 135 cases of the literature. Immunol Res 2017;65:375–85.
[2] Ahn JH, Kim JS, Choi JH, et al. A first case report of pulmonary hyalinizing granuloma associated with immunoglobulin A nephropathy. Medicine (United States) 2017;96:4–7.
[3] Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8–negative/idiopathic multicentric Castleman disease. Blood 2017;129:1646–57.
文字来源:汪劭婷 王琦璞 李霁 张婷 张路 毛玥莹
本文转载自订阅号「协和呼吸」
原链接戳:花季少女,肺内多发团块到底为何(下)| 病例拾萃[21] · 协和呼吸