
 分享
分享
引言
上期分享的案例中(点击前方蓝字可跳转阅读),可以看到患者经历了纷繁复杂的检查过程,在思考为何会在临床诊断与鉴别诊断中遗漏重点的同时,提示大家对原因不明的肺部病变,在诊疗过程中不能仅着眼于肺部病变,而是需要纵观全局,开放思维跳出局限,不放过任何一条异常的检查结果,全面而细致的查体和询问病史。本期接着揭开这个被遗漏的「盲区」,从该案例我们得出的教训是,免疫缺陷病的病因可为原发,也可为继发,前者多为遗传因素,约占60%。原发性免疫缺陷病中的普通变异型免疫缺陷病(common variable immunodeficiency, CVID)患者中可见结节病样病变,有肝脏和肺的肉芽肿浸润,并有不同程度的ACE水平增高。因此,对于看起来像结节病的患者,如出现低丙种球蛋白血症及反复感染,应当评估是否为CVID[1]。
CT引导下肺穿刺,送检组织病理及mNGS;自身免疫性抗体全套;M蛋白;乳胶凝集试验;骨髓穿刺术……这些检查还需完善吗?后续检查结果能否判定病因?
根据现有检查结果,团队进一步讨论患者的可能病因:
1、肺隐球菌病:起病隐匿、进展缓慢,可发生于健康人或免疫受损人群,主要侵犯肺和中枢神经系统。但患者的乳胶凝集试验阴性,目前证据不足,可待肺穿刺病理及特殊染色进一步明确。
2、肺结核:可表现为双肺多发结节样病灶,但多有典型卫星灶、空洞等表现,且分布以肺尖后段、下肺背段为主,患者影像学表现不典型。该患者入院前期复查病灶明显进展,但血沉正常、T-SPOT阴性,无低热、盗汗、消瘦等结核中毒症状,不符合活动性肺结核表现。
3、其他罕见病原体感染:患者白细胞、球蛋白水平低下,结合既往严重带状疱疹,需考虑是否存在罕见病原体感染可能。外院已行气管镜肺泡灌洗及二代测序检查,但考虑当时患者肺部病灶较小且靠近外周,不能排除检测假阴性的可能性。
4、肉芽肿性多血管炎:患者外院多次自身免疫性抗体(包括ANCA)均为阴性,无鼻咽喉等上呼吸道受累表现,无血尿、肾功能减退等肾脏受损表现,无发热等全身症状,不符合GPA。但考虑患者病程长且进展缓慢,不排除病程早期自身免疫性抗体阴性且未累及其他系统的可能。
5、结节病:患者入院后复查胸部增强CT提示双肺多发结节、纵隔淋巴结肿大,符合结节病影像学表现。但患者血沉、血钙、血清血管紧张素转化酶水平均正常,确诊证据仍不足。
6、隐源性机化性肺炎: 对比患者多次胸部CT影像片,患者双肺多发结节样病灶呈游走性,2020-07-14复查双肺部分病灶较2020-04-28明显吸收,此后多次复查胸部CT提示双肺多发结节病灶增多、增大,且较前位置有变化。结合患者目前感染、自身免疫性疾病、结节病等证据均不足,需考虑是否存在隐源性机化性肺炎。
下一步需要完善哪些检查措施?(多选)
- CT引导下肺穿刺,送检组织病理及mNGS
- 自身免疫性抗体全套
- M蛋白
- 乳胶凝集试验
- 骨髓穿刺术
补充检查结果:
1、患者入院后,行CT引导下肺穿刺(左下肺基底段结节),病理:肺肉芽肿性病变,见多个肉芽肿性小结节形成和多核巨细胞,未见干酪样坏死,需结合相关病原学微生物实验室检测,查找病因;免疫组化及特殊染色:CK7(肺泡上皮阳性);TTF-1(肺泡上皮阳性),NapsinA(肺泡上皮阳性),p40(-),CK5/6(+/-),CD68(KP1)(个别阳性),AB(-)/PAS(-),抗酸染色(-),Lysozyme(局灶阳性),六胺银染色(-)。
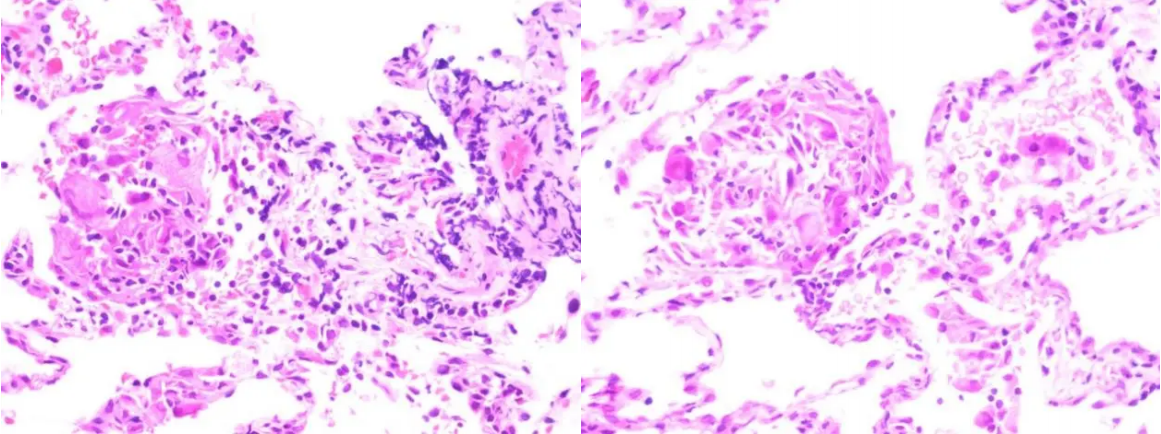
图8:肺穿刺病理(HE染色)
2、mNGS(肺穿刺组织):检出嗜麦芽窄食单胞菌、克雷伯菌等;真菌、结核、支原体、衣原体、病毒未检出。
3、骨髓细胞学:骨髓粒、红、巨三系增生,形态未见明显异常。
4、血检验:
11月11日查自身免疫结果: 抗核抗体:阴性、抗核抗体1:100:阴性、抗核抗体1:1000:阴性、抗核抗体1:10000:阴性、PR3-ANCA:阴性、抗双链DNA(定性):阴性、抗Smith抗体:阴性、抗U1-RNP抗体:阴性、抗SSA抗体:阴性、抗SSB抗体:阴性、抗Jo-1抗体:阴性、抗Scl-70抗体:阴性、MPO-ANCA:阴性、pANCA:阴性、cANCA:阴性、抗肾小球基底膜抗体:阴性、抗组蛋白抗体AHA:阴性、抗核小体抗体:阴性、抗核糖体抗体:阴性、CENP B:阴性、Ro52抗体:阴性、抗双链DNA(定量):1IU/ml;
M蛋白: IgG:0.68g/L↓、IgA:0.28g/L↓、IgM:0.19g/L↓、κ-轻链:0.12g/L↓、λ-轻链:0.11g/L↓、κ/λ比值:1.091↓、IgE:4.46IU/ml,血清蛋白电泳:未见异常蛋白区带。
CD细胞: NK:19.0、NK绝对值计数:169、CD3:77.7%、CD3绝对值计数:690、CD4:47.3%、CD4绝对值计数:420、CD8:27.8%、CD8绝对值计数:247、CD4/CD8:1.70%、CD19:2.1%↓、CD19绝对值计数:18↓。
至此,综合所有检查报告分析结论,可判定患者为双肺肉芽肿性病变。
肺结节穿刺病理得出结论,但回顾长达半年的病程,纷繁复杂的检查结果中是否有线索一直被忽视?
首先分析双肺肉芽肿性病变的病因。肉芽肿性肺疾病(granulomatous lung disease,GLD)是一组以肉芽肿性炎症和肉芽肿形成为共同病理特征的肺部疾病,由于其病因众多、临床表现和胸部影像学不具有特征性,有时病理学也难以明确诊断,故临床诊断和鉴别诊断非常困难,极易造成误诊和漏诊,应引起大家的高度重视。
临床上能引起GLD的病因很多,主要包括:(1)各种微生物感染所致肉芽肿:如结核或非结核分枝杆菌、真菌(如曲霉、隐球菌、组织胞浆菌、球孢子菌)、寄生虫感染等。(2)外源性过敏性肺泡炎。(3)无机粉尘或异物沉积:硅沉着病、铍肺、吸入性肺炎(颗粒性物质吸入) 等。(4)血管炎性肉芽肿病:肉芽肿性多血管炎(GPA)、嗜酸性肉芽肿性多血管炎(EGPA)、淋巴瘤样肉芽肿病、坏死性结节性肉芽肿病、支气管中心性肉芽肿病。(5)结缔组织疾病:类风湿结节。(6)其他原因不明的肉芽肿:结节病、朗格罕斯细胞组织细胞增多症。(7)遗传性:如慢性肉芽肿病、各种先天性免疫缺陷病导致的肉芽肿性淋巴细胞性间质性肺疾病(GLILD)等。通常将GLD分为两大类, 即感染性肺肉芽肿病和非感染性肉芽肿病, 前者最常见的是分枝杆菌和真菌感染, 后者最常见的是结节病和外源性过敏性肺泡炎[1,2]。
患者肺结节穿刺病理提示肺肉芽肿性病变,未见干酪样坏死,特殊染色如AB、PAS、抗酸染色、六胺银染色均为阴性,肺穿刺组织mNGS中真菌、结核均未检出。血检验提示血沉正常,结核T细胞检测阴性;血清GM试验、乳胶凝集试验均为阴性;血钙、血清血管紧张素转化酶正常;自身免疫性抗体全套均为阴性。患者的职业为教师,发病前无特殊接触史。目前感染、过敏性肺炎、职业性肺病、血管炎性肉芽肿病、结缔组织疾病、结节病等诊断均证据不足。回顾患者长达半年的病程,纷繁复杂的检查结果中,是否有线索一直被忽视?
团队又对患者的病史进行了详细回顾,发现应该作以下补充:
1、免疫球蛋白低下:既往血检验提示免疫球蛋白低下,未被重视。
体液免疫(2020.05.07):IgG:1.35g/L↓;IgA:<0.08g/L↓;IgM:0.12g/L↓;IgE:<10IU/ml↓;C3:1.09g/L;C4:0.34g/L;
体液免疫(2020.08.25):IgG:1.01g/L↓;IgA:<0.07g/L↓;IgM:0.09g/L↓;C3:0.91g/L;C4:0.36g/L;Kappa-LC:53.60mg/dl↓;Lambda-LC:59.20mg/dl↓。
2、血小板减少性紫癜:21岁时因四肢紫癜就诊发现血小板低下,自诉发病前无感冒、发热、特殊用药,予糖皮质激素治疗10余天后好转(具体不详)。
3、反复血小板减少:28岁产后反复出现血小板减少,每次均因畏寒、发热就诊,体温高达38-39℃,查血常规提示血小板减少,余无异常,予退热等对症治疗后症状缓解,复查血小板可恢复正常,每年约发生4-5次。
后续诊疗过程:
1、诊断:普通变异型免疫缺陷病(common variable immunodeficiency,CVID)
2、诊断依据:
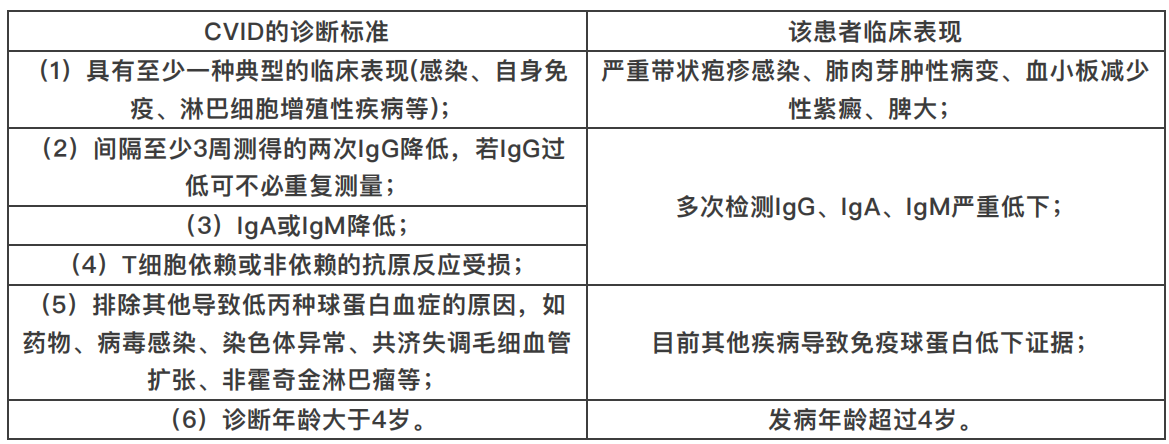
3、治疗及评估:
(1)治疗:予强的松40mg/日抗炎,免疫球蛋白20mg/月替代治疗。
(2)评估:治疗1月后复查胸部CT提示双肺结节灶明显吸收。
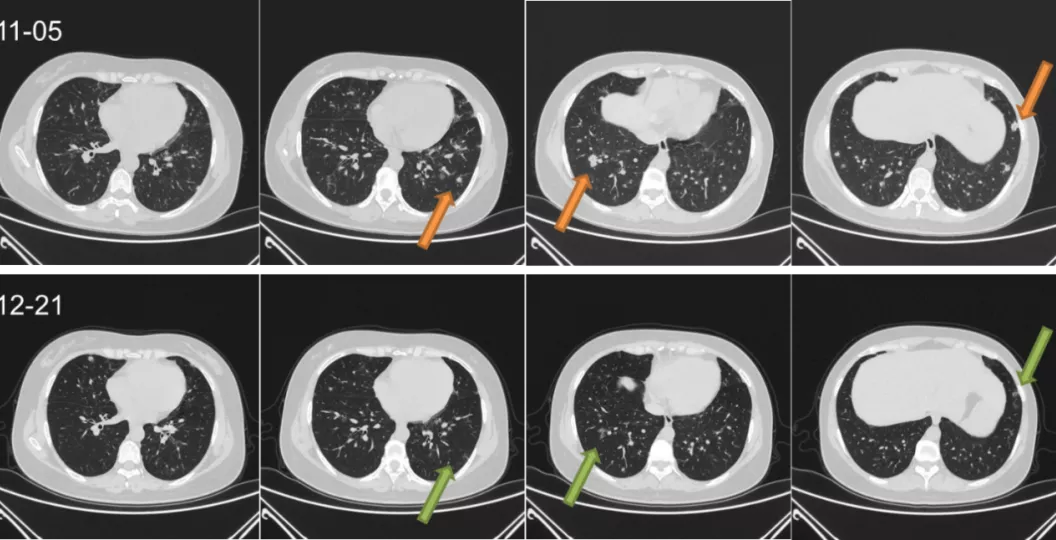
图9:胸部CT对比(2020.11.05、2020.12.21)
入院查体:脾脏肋下可触及,质软。
12月22日检查球蛋白:13g/L、IgG:3.19g/L、IgA:0.28g/L、IgM:0.09g/L、IgE:5.00IU/ml。血常规未见异常。
予免疫球蛋白20mg/月替代治疗,强的松减量至30mg/日出院口服。
后续治疗及随访:
2021年6月,患者因「菌血症(大肠埃希菌)」入院,经治疗后好转。目前强的松减量至15mg/日,间断予免疫球蛋白替代治疗。期间多次复查提示球蛋白、免疫球蛋白IgG、IgA、IgM、CD19+B淋巴细胞比例及计数水平均低下。
重点对普通变异型免疫缺陷病、CVID导致的间质性肺病进行分析,从该案例我们得出哪些经验和教训?
重点对普通变异型免疫缺陷病进行分析:
1、普通变异型免疫缺陷病(common variable immunodeficiency,CVID)是一种原发性免疫缺陷病(primary immunodeficiency,PID)。其特点包括低丙种球蛋白血症、B淋巴细胞分化成熟缺陷、迟发等,需注意该疾病为排除性诊断。
2、临床表现:
(1)感染:反复呼吸道感染、消化道感染等。
(2)非感染性并发症:自身免疫性疾病(如血细胞减少)、肉芽肿性疾病、恶性肿瘤、间质性肺病、克罗恩病、溃疡性结肠炎等。
3、肉芽肿病变:约10%的CVID患者可出现肉芽肿病变,其发病平均年龄为18~34岁。肉芽肿性病变可发生在肺、肝脏、皮肤、胃肠道、脾脏等组织或器官,其中肺为CVID患者肉芽肿性病变最常发器官[3]。

图10:CVID肉芽肿病变累及部位
4、发病机制:CVID的发病机制目前尚不明确。研究认为存在B淋巴细胞成熟分化障碍、抗体产生减少,从而导致低免疫球蛋白血症。约40%-50%的CVID患者外周血B淋巴细胞略减少,约10%的CVID患者B淋巴细胞显著减少或缺失。
5、治疗:主要治疗方法为终生免疫球蛋白替代治疗,建议治疗剂量为每3-4周给予丙种球蛋白0.4-0.8g/kg,尽量保持患者血浆IgG水平在5g/L以上。
重点对CVID导致的间质性肺病进行分析:
1、CVID肺部受累的病理类型:肉芽肿、间质炎症、支气管周围炎症、淋巴细胞浸润、淋巴组织增生、机化性肺炎、纤维化等[4]。因CVID合并的肺间质性病变在病理上表现为特征性的淋巴细胞浸润伴或不伴肉芽肿形成,故称为肉芽肿性淋巴细胞性间质性肺疾病(granulomatous-lymphocytic interstitial lung disease,GLILD)。
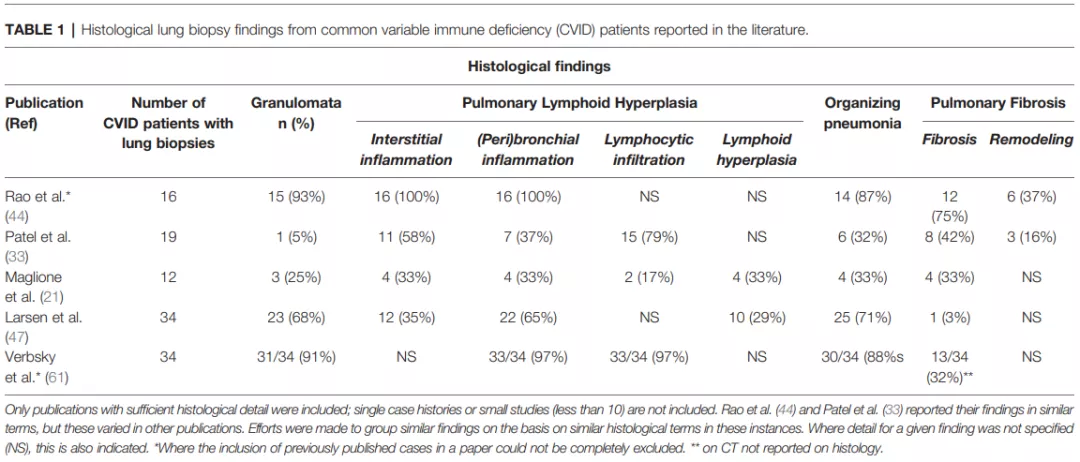
图11:CVID肺部病变的病理类型
2、治疗方案:糖皮质激素单药(最常见)、利妥昔单抗/联合硫唑嘌呤、免疫抑制剂(如MMF、CHX、环孢素等)、TNFα生物制剂等[3]。

图12:CVID肺部肉芽肿病变的治疗方案及疗效比较
3、疗效差异:根据文献报道,糖皮质激素单药的缓解率为66.7%,利妥昔单抗/联合硫唑嘌呤的缓解率为94.4% [3]。
4、糖皮质激素的治疗剂量及疗程:
目前无统一标准,根据欧洲免疫缺陷病协会/欧洲呼吸病协会(ESID/ERS)的网络问卷调查(对象为有GLILD诊疗经验的医生)[5]:
(1)剂量:50%的医生选择强的松1mg/kg,39.0%的医生选择0.5mg/kg。
(2)疗程:46.3%的受访医生选择在1-3月内减至维持剂量或停药。
(3)维持治疗:46.8%的受访医生给予患者糖皮质激素维持治疗。
(4)复发概率:33.8%的受访医生治疗的患者出现复发。
对该案例中得出的经验教训总结如下:
遗传因素和免疫缺陷导致的肺部肉芽肿疾病一直是被忽视的角落。免疫缺陷病的病因可以为原发,也可以为继发,前者多为遗传因素,约占60%。原发性免疫缺陷病中的普通变异型免疫缺陷病(common variable immunodeficiency, CVID)患者中可见结节病样病变,有肝脏和肺的肉芽肿浸润,并有不同程度的ACE水平增高。因此,对于看起来像结节病的患者如出现低丙种球蛋白血症及反复感染,应评估是否为CVID[1]。
肺部病变是多种全身性疾病在人体中的「窗口」,如结缔组织疾病、血管炎、IgG4相关性疾病等。呼吸科医生在原因不明的肺部病变的诊疗过程中,不能仅仅着眼于肺部病变,要纵观全局、开放思维、跳出局限。不放过任何一条异常的检查结果,全面而细致的查体和问病史,有时候能够给我们带来更多的提示。
参考文献 (可上下滑动浏览)
[1]徐作军.应重视对肉芽肿性肺疾病的诊断和鉴别诊断[J].中华结核和呼吸杂志,2020,43(12):1002-1003.
[2]冯瑞娥.做好肉芽肿性肺疾病的病理诊断及鉴别诊断[J].中华结核和呼吸杂志,2020,43(12):1004-1008.
[3]van Stigt AC, Dik WA, Kamphuis LSJ, et al. What Works When Treating Granulomatous Disease in Genetically Undefined CVID? A Systematic Review[J]. Front Immunol, 2020, 11(606389.doi:10.3389/fimmu.2020.606389
[4]Dhalla F, Lochlainn DJM, Chapel H, et al. Histology of Interstitial Lung Disease in Common Variable Immune Deficiency[J]. Front Immunol, 2020, 11(605187.doi:10.3389/fimmu.2020.605187
[5]van de Ven A, Alfaro TM, Robinson A, et al. Managing Granulomatous-Lymphocytic Interstitial Lung Disease in Common Variable Immunodeficiency Disorders: e-GLILDnet International Clinicians Survey[J]. Front Immunol, 2020, 11(606333.doi:10.3389/fimmu.2020.606333
专家介绍

唐昊
上海长征医院(海军军医大学第二附属医院)呼吸与危重症医学科主任,中华医学会呼吸病学分会 9/10届青委会委员,中国医师协会呼吸医师分会青年委员,上海市医学会呼吸病学分会委员,上海市医学会呼吸病学分会哮喘学组副组长,上海市医师协会呼吸医师分会委员,上海市优秀学术带头人,上海市曙光学者,上海市浦江人才,上海市科技启明星。

方正
上海长征医院(海军军医大学第二附属医院)呼吸与危重症医学科副主任医师、副教授,上海市医学会呼吸分会委员,上海市医学会呼吸分会感染学组委员,上海市黄浦区专家组成员。从事呼吸内科工作30多年,擅长肺癌的早期诊断和综合诊治、肺部疑难和危重感染的诊治、慢阻肺和哮喘的治疗,支气管镜的介入治疗、胸膜疾病的胸腔镜诊治等。获军队科技进步二、三等奖各1项。承担国家重大专项基金1项、国家自然科学基金2项、上海市科委基金1项。发表论文30多篇,其中SCI收录8篇;主编专著1部。

宋慧慧
上海长征医院(海军军医大学第二附属医院)呼吸与危重症医学科主治医师、硕士,PCCM 2017级学员。发表科研论文4篇,其中SCI文章2篇,国内核心论文1篇,参与2项国家自然科学基金、1项国家青年科学基金,参与编写专著1本。
本文完
采写编辑:冬雪凝
排版:Jerry