
 分享
分享
摘要
本文系统复习了2021年11月至2022年10月期间国内外权威杂志中关于间质性肺疾病的文献,重点关注了特发性肺纤维化、进展性肺纤维化、结缔组织疾病相关性间质性肺疾病(尤其是类风湿关节炎相关性间质性肺疾病、系统性硬化症相关性间质性间质性肺疾病、特发性炎性肌病相关性间质性肺疾病)、结节病等间质性肺疾病的诊治共识、指南以及其诊疗进展方面的内容。另外,我们还关注了间质性肺疾病的合并症(合并肿瘤、合并肺气肿)方面的内容。
在2022年度国内外在间质性肺疾病(interstitial lung disease, ILD)的诸多领域均进展斐然,尤其是在特发性肺纤维化(idiopathic pulmonary fibrosis, IPF)、进展性肺纤维化(progressive pulmonary fibrosis,PPF),结缔组织疾病相关性间质性肺疾病(connective tissue disease associated ILD,CTD-ILD)、结节病等方面:国际上更新了2015年IPF的诊治指南[1],并首次提出了PPF的诊断标准和治疗推荐[1];发表了多个涉及国际多中心的IPF、结节病、类风湿关节炎相关性ILD、系统性硬化症相关性ILD等的药物研究结果[2-8];在炎性肌病相关性ILD方面则重点关注了不同区域的抗黑色素瘤分化相关基因5(Anti melanoma differentiation associated gene 5 )相关性ILD的临床特点[9-12];中国专家们提出了我国CTD-ILD的诊疗规范、特发性炎性肌病相关性ILD的专家共识、抗肿瘤药物相关间质性肺病诊治专家共识等多个ILD方面的纲领性文件[13-15]。由于篇幅有限,本年度综述未能纳入2022年度ILD方面的所有卓越文献。
一、特发性肺纤维化诊疗进展
国内外学者分享了进行中的IPF研究的内容,使大家能更好的了解不同地域的IPF特征。在IPF流行病学方面,1.07千万美国退伍军人中,从2010年至2019年IPF明显增多:IPF患病率由2010年的276/10万增加到2019年的725/10万,其中2010年诊断IPF为73/10万、2019年则为210/10万;经统计学分析,发现白种人、吸烟、居于农村是他们发生IPF的危险因素[16]。澳大利亚的数据则表明,IPF的发病率为10.4~11.2/10万、患病率为32.6~35.1/10万、死亡率为5.9~6.3/10万;虽然70岁以上的IPF患者仅占9%,但其占死亡患者中的82-83%[17]。
在IPF诊断方面,芝加哥大学的Onishchenko等人研发了一套基于患者电子病历中共病信息的早期诊断IPF的评分体系——零负担共病风险评分-IPF(zero-burden comorbidity risk score for IPF,ZCoR-IPF);零负担是指不需要额外的辅助检查和临床问诊[18]。并经独立数据库的大数据验证,用该评分体系在常规方法诊断IPF前1年和4年预测IPF诊断的ROC曲线下面积分别为0.88和0.84,有望在临床工作中推广本评分体系以早期诊断IPF、早期接受针对性治疗而改善预后。为了能更准确的临床诊断IPF、更少的安排纤维化性ILD患者外科肺活检,Cottin等提出了基于胸HRCT和/或肺活检病理特征,整合是否具备IPF相关临床信息和疾病行为相关信息,使用阶梯贝叶斯方法对IPF相关的临床信息和疾病行为进行汇总分析,从而更精确地判断纤维化性ILD:支持IPF诊断的临床信息包括“男性”、“>60岁”、“有吸烟史”、“爆裂音”、“杵状指”、“无风湿免疫色彩”、“有纤维化性ILD家族史”、“限制性通气功能障碍”以及“慢性起病”;支持IPF诊断的疾病行为相关信息包括“即便接受治疗病情仍在数月或数年内进展”、“对激素和/或免疫抑制剂无效”、“支气管肺泡灌洗液中淋巴细胞比例不高(<15%)”、“无IPF相关的基因学特征”、“经支气管镜肺活检标本的Envisia基因组分析不支持UIP表现”[19-20]。
Jegal等报道了韩国自2008年以来的2139例IPF患者[21]:平均年龄(67.4±9.3)岁,男性占76.1%,71.0%有吸烟史,首诊时无症状者占14.4%,56.9%病例的GAP评分为I级;2016年以来,IPF患者中接受抗纤维化药物治疗(>3月)占79.2%,其中有62.4%的IPF患者接受了吡非尼酮治疗;高龄、IPF急性加重、未接受抗纤维化药物以及有木屑或矿尘接触史是IPF患者死亡相关因素。美国IPF转归前瞻性队列研究(IPF-PRO)发现,收入较高、就诊于具肺移植资质的医疗中心的IPF接受肺移植的概率更高[22]。我们的回顾性分析发现,IPF患者若出现发热、关节肿痛等关节炎表现以及镜下血尿时,要警惕类风湿关节炎和显微镜下多血管炎等CTD-UIP的可能性;在诊断CTD-UIP之前的“IPF”阶段抗纤维化可获益,在诊断CTD-UIP后建议激素联合免疫抑制剂治疗[23]。
IPF治疗探索方面,磷酸二酯酶4B(phosphodiesterase 4B,PDE4B)抑制剂BI1015550相对于安慰剂治疗IPF 12周的II期临床试验结果显示[6]:基线无抗纤维化药物的IPF患者,BI1015550组vs安慰剂组,FVC变化的中位数为5.7 ml (95%CI: 39.1~50.5) vs–81.7 ml (95%CI: 133.5~–44.8),两组相差88.4ml(95%CI: 29.5~154.2);基线已接受抗纤维化药物的IPF患者,BI1015550组vs安慰剂组,FVC变化的中位数为2.7ml(95%CI:–32.8~38.2)vs–59.2 ml (95%CI:–111.8~–17.9),两组相差62.4ml(95%CI:6.3~125.5)。两组的不良事件以及严重不良事件类似。但抗αvβ6的IgG1型单抗(BG00111)并未能有效治疗IPF[4]:中期分析(26周)发现BG00011组vs安慰剂组FVC的最小平方均值为-0.097L vs -0.056L(p=0.268),BG00011组IPF病情更重:更多患者在BG00011组出现胸CT上肺纤维化加重(44.4% vs 18.2%)、IPF加重(13例 vs 0)以及重症不良事件。国际多中心的RCT研究表明,色甘酸钠吸入治疗也未能有效缓解IPF患者的慢性咳嗽[2]。比较糖皮质激素联合静脉环磷酰胺vs糖皮质激素治疗IPF急性加重的法国III期多中心临床试验表明[3]:糖皮质激素联合环磷酰胺组3月的全因死亡率高于单药糖皮质激素组(45% vs 31%)。
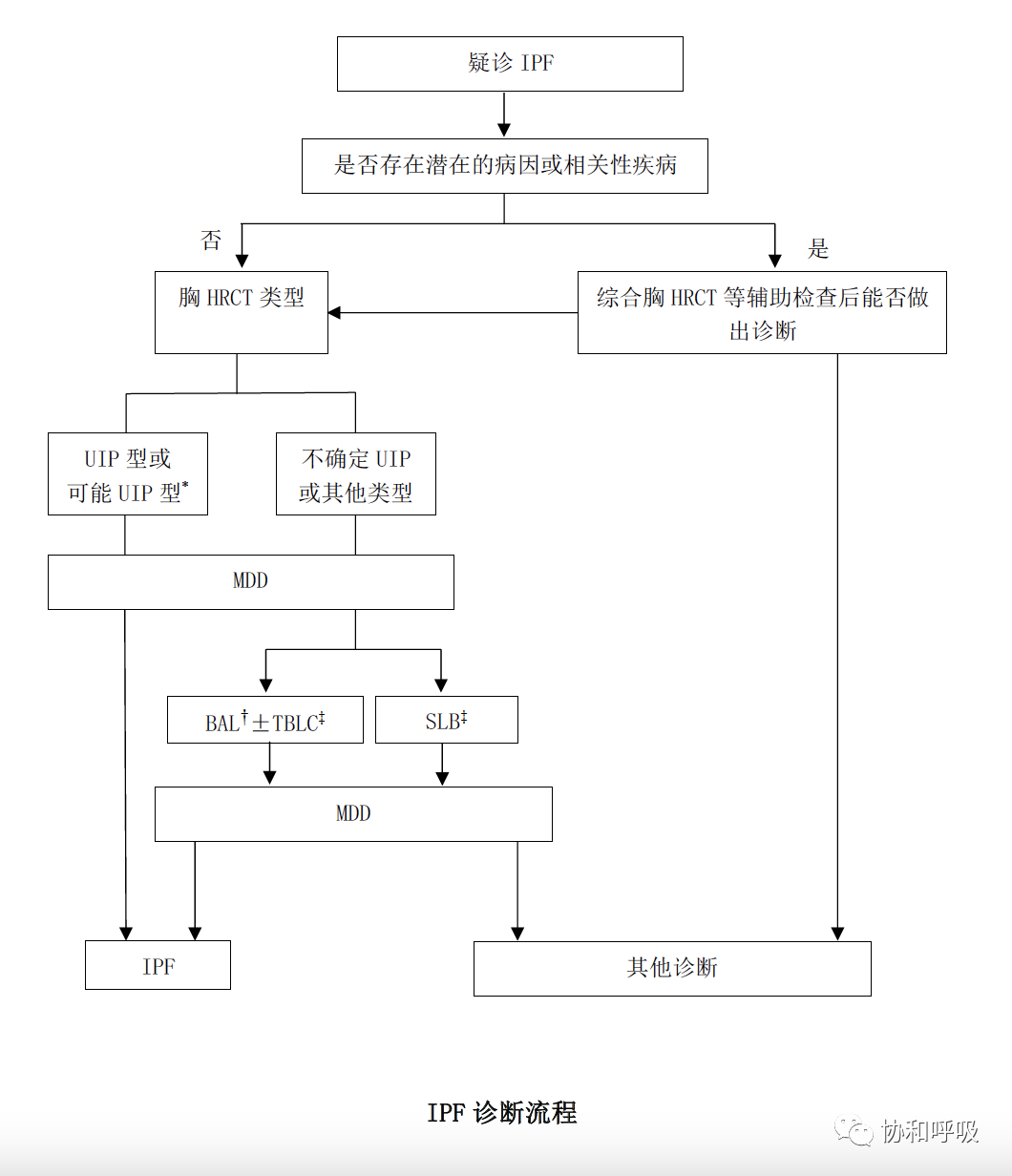
国际专家在2022年5月更新了IPF的临床诊疗指南[1, 24],主要更新点在于:在IPF诊断流程中:将胸HRCT的可能UIP型等同于UIP型对待,即可能UIP型的也未必要安排肺活检,综合临床特征、化验等资料经多学科讨论后诊断IPF;综合胸HRCT表型和肺组织活检病理表型时,CT显示为非UIP型患者,若经肺组织活检,病理提示可能UIP型则诊断不确定的IPF(在2018版的指南中认为是非IPF)。提出了基于循证证据的诊断IPF的专家推荐意见:由有经验的专家操作经支气管镜冷冻肺活检获取肺组织、并请有经验的病理专家解读时,支气管镜冷冻肺活检可替代外科肺活检用于形态学表型未明的ILD是否为UIP的鉴别诊断中;尚无足够的证据来支持或反对通过基因检测协助经支气管镜钳取活检后、形态学表型仍未明的ILD是否为UIP的鉴别诊断。基于循证证据的IPF治疗推荐:不推荐将抑酸药物、胃-食管抗反流的外科手术作为改善IPF预后的治疗措施[1, 24]。
二、结缔组织疾病相关性间质性肺疾病诊疗进展
CTD-ILD是近几年来ILD领域进展很大的领域,本综述重点关注了类风湿关节炎相关性ILD、系统性硬化症相关性ILD、特发性炎性肌病相关性ILD等常见CTD-ILD的诊疗进展。
首个特发性炎性疾病相关性ILD的中国专家共识,全面提出了这类CTD-ILD的诊断、治疗及随访策略[13]:建议胸HRCT、肺功能、多学科讨论的诊疗模式用于首诊、随诊中,首诊中推荐检测肌炎抗体谱,不建议常规开展肺活检,但若需要与CTD-ILD鉴别则可以开展肺活检。建议结合ILD的严重程度、起病形式、进展方式以及肌炎特异性抗体等来综合评价后制定治疗方案,对于初治、复发、难治性、快速进展性、MDA-5相关性ILD等不同类型的炎性肌病ILD分别给出了专家推荐方案,并推荐出现进展性肺纤维化表型的炎性肌病ILD加用抗纤维化药物治疗[14]。中华医学会风湿病学分会则推出了CTD-ILD的诊疗规范,提出了分别从CTD和ILD两方面来全面评价、规范化诊疗CTD-ILD以达到双重达标;此外,还重点介绍了类风湿关节炎相关性ILD、系统性硬化症相关性ILD、特发性炎性肌病相关性ILD的管理策略。
抗MDA-5抗体相关性ILD仍是炎性肌病相关性ILD中非常瞩目的ILD:武雅楠等比较了抗MDA-5抗体阳性与阴性的特发性炎性肌病患者的临床特征,发现抗MDA-5抗体阳性患者的皮肌炎典型皮肤损害多见,但肌肉症状轻,合并ILD比例高、预后差。上海仁济医院的Lian等报道的中国MDA-5-ILD长期随诊队列(216例),发现这些患者即便接受了积极治疗死亡率(39.3%)仍很高,但是一旦他们活过第一年,经治疗后其在血清标记物和肺功能指标方面都有很大的改善[9]。南京的队列分析(272例)也发现,病程短、高水平的血清CRP及抗MDA-5滴度、抗Ro-52阳性是皮肌炎患者发生快速进展性ILD的危险因素;诊断抗MDA-5相关性ILD的头3月是死亡的高发期,50%的快速进展性抗MDA-5相关性ILD及46%的抗MDA-5阳性的患者均在这段时间内死亡,而一旦活过1年则发生快速进展性ILD和死亡时间均明显下降[11]。国际多中心的非亚裔抗MDA-5阳性的CTD回顾性队列(149例)分析发现,78%(108例)的患者合并ILD,21.5%(32例)的患者出现快速进展性ILD(其中19例死亡,本队列中共有26例患者死亡),13%(20例)患者合并恶性肿瘤;他们建议风湿科、呼吸科以及ICU专家组成团队来共商此类患者的救治,以期改善其预后[10]。
国际上首个显微镜下多血管炎的分类标准出炉,指出对于中或小血管炎的患者,除外感染性疾病等类似血管炎的疾病后,若评分标准得分≥5分便可诊断显微镜下多血管炎,具体标准如下:减分项目:鼻部病变(-3)、外周血嗜酸粒细胞≥1×109/L(-4)、cANCA或抗蛋白酶3抗体阳性(-1);加分项目:pANCA或抗过氧化物酶抗体阳性(+6)、胸部影像学提示肺纤维化或间质性肺疾病(+3)、肾活检提示少免疫性肾小球肾炎(+3),从而进一步简化了显微镜下多血管炎的诊断标准[25]。我国的吴挺挺等发现显微镜下多血管炎相关性ILD患者全身血管炎症状较少,大部分ILD出现在血管炎诊断之前或诊断时,ILD以UIP型或UIP样为主, LDH建议尽早使用激素联合免疫抑制剂或利妥昔单抗以改善预后;发现血清乳酸脱氢酶升高是生存期缩短的独立危险因素,伴有血清类风湿因子升高则是生存改善的保护因素[26]。
托珠单抗治疗系统性硬化症相关性ILD的延长期研究表明,无论是新近启用组(之前为安慰剂组)还是持续使用组(之前为托珠单抗组)均能有效的延缓这类患者肺功能FVC的下降,且长期安全性良好[8]。相对于接受环磷酰胺治疗组而言,造血干细胞移植组的系统性硬化症相关性ILD患者在上述治疗后1年时复查的胸CT所示,ILD相关影像表现有所改善(但差异无统计学意义),可能还需要延长随诊观察年限来评价干细胞移植组的疗效[7]。首个全球多中心的吡非尼酮vs安慰剂治疗类风湿关节炎-ILD的随机、对照研究共筛选231例患者,最终入组123例(吡非尼酮组vs安慰剂组=63例:60例)(排除标准:吸烟史、其他原因所致的ILD、合并有明显临床表现的COPD、哮喘患者)。本研究为期52周,未达成主要研究终点(FVC%预计值下降≥10%或死亡)[27]:吡非尼酮组vs 安慰剂组=11%(7/63)vs 15%(9/60)[OR:0.67,95%CI:0.22~2.03,p=0.48]。但关键的次要终点指标——FVC的年绝对下降值及FVC%预计值的下降值在两组间均有显著性差异:吡非尼酮组vs 安慰剂组=66ml vs 146ml(p=0.0082),吡非尼酮组vs 安慰剂组=1.02% vs 3.21(p=0.0028)。两组之间的用药安全性方面无统计学差异。
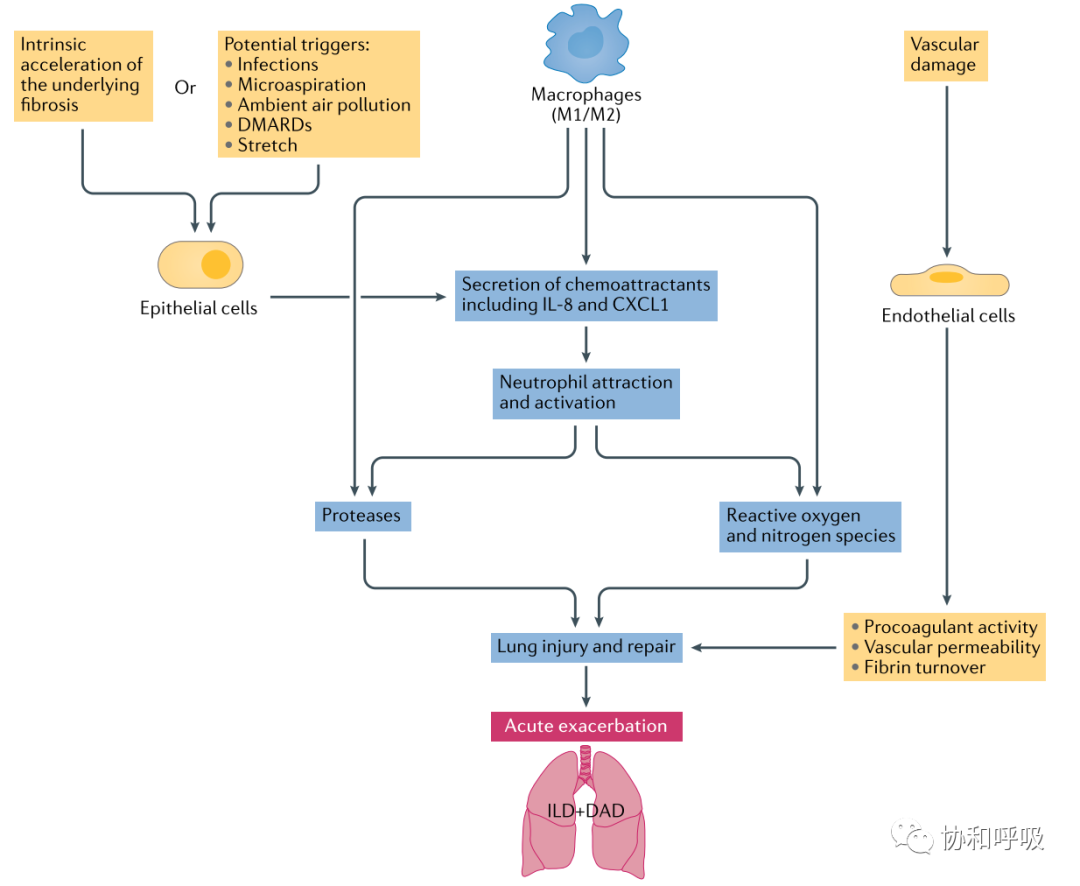
Luppi等首次提出了风湿免疫病相关性ILD(RD-ILD)急性加重(AE-RD-ILD)的概念,并详细论述了其危险因素、临床表现、诊断和治疗推荐[28]:UIP型的RD-ILD均可能出现类似IPF急性加重事件的AE-RD-ILD,以类风湿关节炎相关性ILD、系统性硬化症重叠肌炎或皮肌炎相关性ILD、系统性血管炎相关性ILD多见,感染、微误吸、空气污染、机械牵张损伤、DMARDs药物等是常见的诱因,AE-RD-ILD诊断是一除外性诊断,治疗方案则建议参照IPF急性加重的治疗策略;并呼吁开展更多高级别的临床研究以期为提供基于循证的治疗推荐提供依据。类风湿关节炎相关性ILD急性加重是影响类风湿关节炎预后的重要事件[29-31]。Kwon等发表的类风湿关节炎相关性ILD急性加重的回顾性研究表明,近1/3(28%)的患者发生急性加重,既往吸烟史、基线肺功能差以及活动耐量差是这类患者发生急性加重时间的危险因素,而一旦发生急性加重,严重影响类风湿关节炎相关性ILD的生存(急性加重或30天和90天的死亡率高达12.6%、29.9%)[30]。Otsuka等则发现低DLCO%以及低白蛋白血症是风湿关节炎相关性ILD急性加重患者预后不良的因素,不过,虽然类风湿关节炎相关性ILD急性加重是类风湿关节炎相关性ILD的主要死亡原因,但类风湿关节炎相关性ILD急性加重患者的预后要比IPF急性加重患者好(急性加重时间后的中位生存时间277天vs60天,p=0.038)[31]。
三、进展性肺纤维化和结节病
今年5月发布了进展性肺纤维化(PPF)的首个国际指南,明确提出此版指南中的PPF不包括IPF,PPF是指一大类病因已知或未知的、胸CT上有明确的肺纤维化,在近1年内出现病情进展≥2项如下表现的非IPF-FILD患者:1)咳嗽、气短等呼吸系统症状恶化;2)肺功能下降:1年内FVC绝对值下降>5%,和/或1年内DLco(经血色素校正后)绝对值下降>10%;3)胸部影像学进展(≥1项):牵张性支气管扩张/细支气管扩张范围增大或程度加剧;新发磨玻璃影伴有牵张性细支气管扩张;新发细网格影;粗网格影范围增大或程度加剧;新发蜂窝影或原有蜂窝影加剧;肺容积进一步缩小。并提出基于循证证据的非PPF的治疗推荐:在非IPF-FILD患者经标准治疗失败、呈现为PPF时,推荐使用尼达尼布(有条件的推荐,低证据级别);推荐开展临床试验来进一步评价尼达尼布治疗特殊的非IPF-PPF的有效性、安全性;推荐开展临床试验来进一步评价吡非尼酮治疗一般的非IPF-PPF、特殊的非IPF-PPF的有效性、安全性[1, 24]。
荷兰专家发布了非IPF-进展性纤维化性间质性肺疾病(progressive fibrosing interstitial lung diseases, PF-ILD)诊疗指南(以提问-回答的模式,包括6条诊断方面的推荐意见,8条治疗方面的推荐意见),对于临床工作也很有指导价值[32]:非IPF-FILD在24月内出现临床表现、肺功能和胸部影像学加重时考虑PF-ILD,具体诊断标准为:①FVC%绝对值下降≥10%预计值;或②FVC%绝对值下降5-10%预计值,并DLco%绝对值下降>15%预计值或临床症状加重或胸CT所示病变加重;或③临床症状加重并胸CT所示病变加重。专家组建议对每个FILD患者均尽可能明确其潜在的病因诊断,应对其常规筛查血清自身抗体检测、获取每个FILD患者多学科讨论后的诊断;对不符合PF-ILD诊断标准的FILD患者也要进行规律随诊,以期早期发现疾病进展;建议对于所有的系统性自身免疫性疾病的患者常规进行是否合并ILD的筛查,并建议多学科讨论团队来诊疗CTD-ILD患者。每位PF-ILD患者必须接受针对潜在的FILD的一线治疗药物;若经上述治疗后疗效欠佳,建议加用尼达尼布;但是否加用吡非尼酮则无支持或反对的推荐意见。对于UIP型或纤维化型NSIP型的PF-ILD患者,若因潜在的FILD不必要接受激素和/或免疫抑制剂治疗时,建议抗纤维化药物作为一线治疗药物。抗纤维化药物和激素和/或免疫抑制剂治疗可以联合起来用于需要抗炎治疗的PF-ILD患者。专家们对于是否停用即便接受了抗纤维化药物治疗、但病情仍进展的PF-ILD不做推荐(停或不停均可)。在IPF指南中推荐的非药物治疗措施和肺移植对于PF-ILD患者同样使适用。
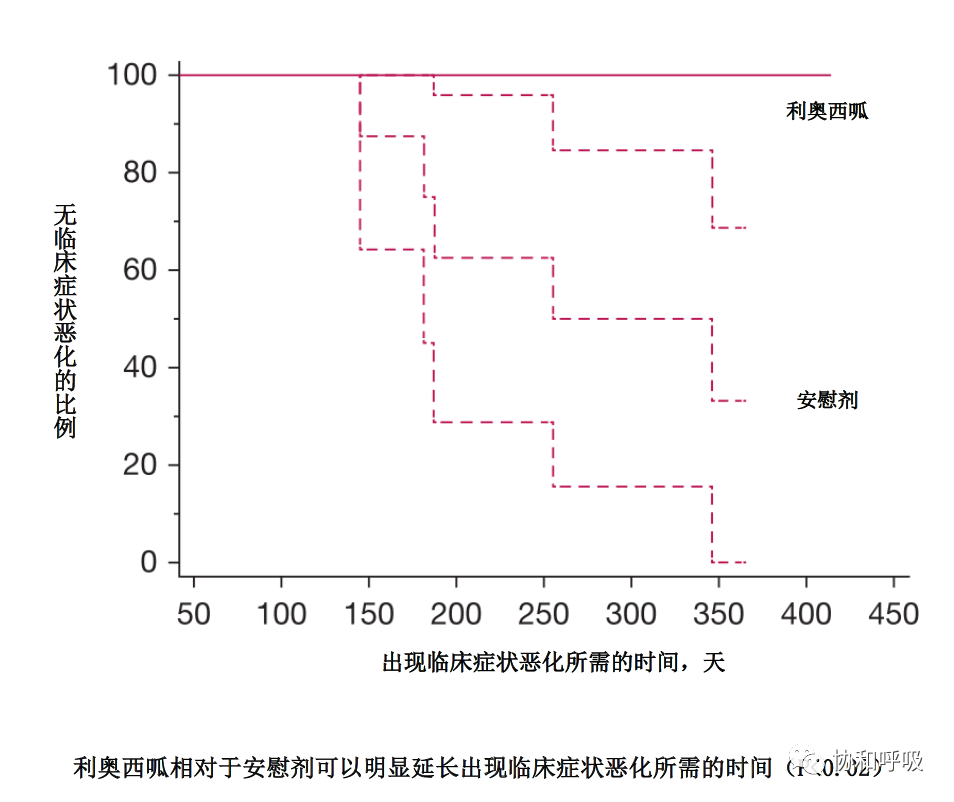
JAMA杂志系统性介绍了肺结节病的规范化诊断流程和治疗策略,尤其是对于激素、免疫抑制剂及生物制剂的使用的推荐等级;此外还简要介绍了Lofgren综合征和结节病并肺高压的相关内容[33],堪为教科书式的肺结节病诊疗的文献。邵池等报道了中国的几例以心律失常起病的结节病患者[34]:提出对于不明原因的高度房室传导阻滞、室性心律失常的中年患者,需要考虑心脏结节病的可能;对于此类患者可以进一步安排PET-CT、心肌增强核磁等检查来辅助诊断;一旦诊断,建议激素联合免疫抑制剂治疗。Baughman等发现,应用一年及以上利奥西胍可以使结节病性肺高压患者的活动耐量改善、阻止其临床症状的恶化[5]。Gupta等则通过综述系统性介绍了进展性结节病的定义、诊断和治疗[35]:进展性结节病是指结节病所累及的脏器功能丧失或导致死亡,进展性肺结节病则特指结节病性肺纤维化、支气管扩张和肺高压,肺部感染(尤其是慢性肺曲霉菌病)、是结节病肺纤维化、支气管扩张的常见并发症。若泼尼松10mg/d仍未能有效治疗结节病支气管扩张,建议加用免疫抑制剂、抗肿瘤坏死因子制剂、利妥昔单抗或JAK抑制剂等;肺纤维化患者则可以尝试加用吡非尼酮、尼达尼布等抗纤维化药物;肺高压患者则需要根据不同类型的肺高压针对性治疗。终末期的结节病肺纤维和/或肺高压患者,可以考虑肺移植。
四、其他
不少学者还关注了ILD合并症,比如恶性肿瘤、肺气肿等常见情况。欧洲的多国、多中心IPF注册登记研究发现[36]:绝大部分(91.3%)IPF患者(3680例)都有共病,37.8%的IPF还合并≥4个共病,其死亡风险较无共病的IPF患者增加44%。死亡风险增随着共病数目增加而增加,即便接受抗纤维化药物治疗也是如此;共病是其中26.1%患者的死因,所以要高度关注IPF患者的共病情况。医科院肿瘤医院的马飞等提出了抗肿瘤药物相关性间质性肺疾病诊治中国专家共识[15],建议由肿瘤科、呼吸科、放射科、病理科和药剂科医师组成的多学科讨论团队来一起指导这类患者的诊断、治疗和长程管理,并提出了这类ILD患者的诊疗路径、基于CTCAE的病情程度分级来制定分级管理策略,建议要识别高风险人群、关注ILD的早期症状和体征,尽早安排胸CT等辅助检查以早期诊断、早期治疗。
肺纤维化并肺气肿综合征(CPFE)的首个国际共识提出了CPFE的定义、临床特征、肺功能特点、共病以及未来的研究内容等[37]:CPFE是一个综合征,而非一种疾病;CPFE中的间质性肺病主要见于IPF、CTD-ILD、过敏性肺炎、各种职业性肺病(如石棉肺、硅肺)等。以吸烟、男性患者多见,但部分CTD-ILD患者和职业性肺病患者可以无吸烟史。临床以咳嗽、活动后气短为主要表现;通气功能保留或轻度异常(FV1/FVC一般正常或轻度下降),但弥散功能明显下降是其肺功能损害的特点(FVC/DLCO一般较高,相较于IPF患者,CPFE患者的FVC更高、DLCO更低)。CPFE的肺脏病理报道相对有限,常常合并有吸烟相关性ILD的特点(呼吸性细支气管炎、吸烟相关性间质纤维化);定量CT对于CPFE患者的肺内病变程度评价非常重要。肺癌和肺高压是常见的共病,基线存在IPF等纤维化性间质性肺疾病时也会出现急性加重的过程。专家组提出了基于不同用途的CPFE的诊断标准,即供CPFE研究的诊断标准和供临床诊疗用的诊断标准。现阶段,CPFE的治疗是综合治疗措施,包括戒烟、针对基线ILD和肺气肿的治疗、针对合并症的治疗以及肺康复策略。最后从CPFE的流行病学、生物病理学、诊断、处理及临床试验等多方面、多角度提出了尚需要进一步探究的研究方向和问题。
IPF新药探索、CTD-ILD领域、ILD合并症等方面的进展依然将会是2023年ILD年度综述中的重要内容。而随着PPF、CPFE等国际共识中相关研究的进行,未来几年将会有PPF、CPFE的诊断、治疗、预后、生物标记物等多方面的内容更新。
作者简介

黄慧
北京协和医院呼吸与危重症医学科主任医师,教授,博士生导师、博士后导师。
参考文献 (可上下滑动浏览)
1. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline[j]. Am J Respir Crit Care Med, 2022,205(9):e18-47. DOI: 10.1164/rccm.202202-0399ST
2. Martinez FJ, Wijsenbeek MS, Raghu G, et al. Phase 2B study of inhaled RVT-1601 for chronic cough in idiopathic pulmonary fibrosis: A multicenter, randomized, placebo-controlled study (SCENIC Trial)[J]. Am J Respir Crit Care Med, 2022,205(9):1084-1092. DOI:10.1164/rccm.202106-1485OC
3. Naccache JM, Jouneau S, Didier M, et al. Cyclophosphamide added to glucocorticoids in acute exacerbation of idiopathic pulmonary fibrosis (EXAFIP): a randomised, double-blind, placebo-controlled, phase 3 trial[J]. Lancet Respir Med, 2022,10(1):26-34. DOI:10.1016/S2213-2600(21)00354-4
4. Raghu G, Mouded M, Chambers DC, et al. A phase IIb randomized study of an anti-αvβ6 monoclonal antibody in idiopathic pulmonary fibrosis[J]. Am J Respir Crit Care Med, 2022,206(9):1128-1139. DOI: 10.1164/rccm.202112-2824OC
5. Baughman RP, Shlobin OA, Gupta R, et al. Riociguat for sarcoidosis-associated pulmonary hypertension: Results of a 1-year double-blind, placebo-controlled trial[J]. Chest, 2022;161(2): 448-457. DOI:10.1016/j.chest.2021.07.2162
6. Richeldi L, Azuma A, Cottin V, et al. Trial of a preferential phosphodiesterase 4B inhibitor for idiopathic pulmonary fibrosis[J]. N Engl J Med, 2022;386(23):2178-87. DOI:10.1056/NEJMoa2201737
7. Ciaffi J, van Leeuwen NM, Boonstra M, et al. Evolution of systemic sclerosis–associated interstitial lung disease one year after hematopoietic stem cell transplantation or cyclophosphamide[J]. Arthritis Care Res, 2022;74(3):433-441. DOI: 10.1002/acr.24451.
8. Khanna D, Lin CJF, Furst DE, et al. Long-term safety and efficacy of tocilizumab in early systemic sclerosis-interstitial lung disease: Open-label extension of a phase 3 randomized controlled trial[J]. Am J Respir Crit Care Med, 2022;205(6):674-684. DOI:10.1164/rccm.202103-0714OC
9. Lian XY, Ye Y, Zou J, et al. Longitudinal study of patients with antimelanoma differentiation-associated gene 5 antibody-positive dermatomyositis-associated interstitial lung disease[J]. Rheumatology, 2022:kaec525. DOI:10.1093/rheumatology/keac525
10. Cavagna L, Meloni F, Meyer A, et al. Clinical spectrum time course in non-Asian patients positive for anti-MDA5 antibodies[J]. Clin Exp Rheumatol, 2022;40(2):274-283. DOI:10.55563/clinexprheumatol/di1083
11. You H, Wang L, Wang J, et al. Time-dependent changes in RPILD and mortality risk in anti-MDA5+ DM patients: a cohort study of 272 cases in China[J]. Rheumatology, 2022;keac450. DOI:10.1093/rheumatology/keac450
12. 王佳佳, 王磊, 徐凌霄, 吕成银, 朱玉静. HIS评分在抗黑色素瘤分化相关基因5抗体阳性皮肌炎合并间质性肺疾病预后评估中的应用价值[J]. 中华风湿病学杂志, 2022;26(4):224-230. DOI: 10.3760/cma.j.cn141217-20210830-00350
13. 邹庆华, 路跃武, 周京国, 刘晓霞, 李梦涛, 赵岩. 结缔组织疾病相关间质性肺疾病诊疗规范[J]. 中华内科杂志, 2022;61(11):1217-1223.
DOI: 10.3760/cma.j.cn112138-20220525-00406
14. 中国研究型医院学会呼吸病学专委会. 特发性炎性肌病相关间质性肺疾病诊断和治疗中国专家共识[J]. 中华结核和呼吸杂志, 2022;45(7):635-650. DOI: 10.3760/cma.j.cn112147-20220108-00025
15. 抗肿瘤药物相关间质性肺病诊治专家共识专家委员会. 抗肿瘤药物相关性间质性肺病诊治专家共识[J]. 中华肿瘤杂志, 2022;44(7):693-702. DOI: 10.3760/cma.j.cn112152-20220412-00244.
16. Kaul B, Lee JS, Zhang N, et al. Epidemiology of idiopathic pulmonary fibrosis among U.S. veterans, 2010-2019[J]. Ann Am Thorac Soc, 2022;19(2):196-203. DOI:
10.1513/AnnalsATS.202103-295OC
17. Cox IA, Otahal P, de Graaff B, et al. Incidence, prevalence and mortality of idiopathic pulmonary fibrosis in Australia[J]. Respirology, 2022;27(3):209-216. DOI:
10.1111/resp.14194
18. Onishchenko DM Marlowe RJ, Ngufor CG, et al. Screening for idiopathic pulmonary fibrosis using comorbidity signatures in electronic health records[J]. Nat Med, 2022;28(10):2107-2116. DOI:10.1038/s41591-022-02010-y.
19. Richeldi L, Scholan MB, Lynch DA, et al. Utility of a molecular classifier as a complement to high-resolution computed tomography to identify usual interstitial pneumonia[J]. Am J Respir Crit Care Med, 2021;203(2):211-220. DOI: 10.1164/rccm.202003-0877OC
20. Cottin V, Tomassetti S, Valenzuela C, et al. Integrating clinical probability into the diagnostic approach to idiopathic pulmonary fibrosis: An international working group perspective[J]. Am J Respir Crit Care Med, 2022;206(3):247-259. DOI:10.1164/rccm.202111-2607PP.
21. Jegal Y, Park JS, Kim SY, et al. Clinical features, diagnosis, management, and outcomes of idiopathic pulmonary fibrosis in Korea: Analysis of the Korea IPF Cohort (KICO) Registry[J]. Tuberc Respir Dis. (Seoul), 2022;85(2):185-194. DOI:
10.4046/trd.2021.0123.
22. Swaminathan AC, Hellkamp AS, Neely ML, et al. Disparities in Lung Transplant among Patients with Idiopathic Pulmonary Fibrosis: An Analysis of the IPF-PRO Registry. Ann Am Thorac Soc, 2022;19(6):981-990.
DOI: 10.1513/AnnalsATS.202105-589OC.
23. 陈茹萱, 刘湘宁, 邵池, 等. 以特发性肺纤维化起病的自身免疫性疾病相关性间质性肺疾病患者临床特征分析[J]. 中华结核和呼吸杂志, 2022;45(8):775-782. DOI: 10.3760/cma.j.cn112147-20220417-00327
24. 黄慧, 徐作军. 国际特发性肺纤维化指南及进展性肺纤维化临床诊疗指南摘译[J]. 中华结核和呼吸杂志, 2022;45(7):721-724. DOI: 10.3760/cma.j.cn112147-20220501-00368
25. Suppiah R, Robson JC, Grayson PC, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis[J]. Ann Rheum Dis, 2022;81(3):321-326. DOI:
10.1136/annrheumdis-2021-221796.
26. 吴挺挺, 岑泽凯, 周海军, 等. 显微镜下多血管炎相关间质性肺疾病28例临床特征及生存分析[J]. 中华结核和呼吸杂志, 2022;45(10):1022-1030. DOI: 10.3760/cma.j.cn112147-20220208-00097
27. Solomon JJ, DanoffSK, Woodhead FA, et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: a randomised, double-blind, placebo-controlled, phase 2 study[J]. Lancet Respir Med,2022;S2213-2600(22):00260-0. DOI: 10.1016/S2213-2600(22)00260-0
28. Luppi F, Sebastiani M, Salvarani C, Bendstrup E, Manfredi A. Acute exacerbation of interstitial lung disease associated with rheumatic disease[J]. Nat Rev Rheumatol, 2022;18(2):85-96. DOI:10.1038/s41584-021-00721-z.
29. Hozumi H, Kono M, Hasegawa H, et al. Acute exacerbation of rheumatoid arthritis-associated interstitial lung disease: mortality and its prediction model[J]. Respir Res, 2022;23(1):57. DOI:10.1186/s12931-022-01978-y.
30. Kwon BS, Lee HY, Choe J, Chae EJ, Hong S, Song JW. Acute respiratory deterioration in rheumatoid arthritis-associated interstitial lung disease: A single-center study[J]. Chest, 2022;162(1):136-144. DOI:10.1016/j.chest.2022.01.007
31. Otsuka J, Yoshizawa S, Kudo K, et al. Clinical features of acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: Comparison with idiopathic pulmonary fibrosis[J]. Respir Med, 2022;200:106898. DOI:10.1016/j.rmed.2022.106898
32. Piotrowski WJ, Martusewicz-Boros MM, Bialas AJ, et al. Guidelines of the Polish Respiratory Society on the diagnosis and treatment of progressive fibrosing interstitial lung diseases other than idiopathic pulmonary fibrosis[J]. Adv Respir Med, 2022;90(5):425-450. DOI:10.3390/arm90050052.
33. Belperio JA, Shaikh F, Abtin FG, et al. Diagnosis and treatment of pulmonary sarcoidosis: A review[J]. JAMA, 2022;327(9):856-867. DOI:10.1001/jama.2022.1570
34. 邵池, 孙宇新, 于琛, 等. 以心律失常起病的结节病的临床特征分析[J]. 中华结核和呼吸杂志, 2022;45(2):183-190. DOI:10.3760/cma.j.cn112147-20210609-00408.
35. Gupta R, Judson MA, Baughman RP. Management of advanced pulmonary sarcoidosis[J]. Am J Respir Crit Care Med, 2022;205(5):495-506.
DOI: 10.1164/rccm.202106-1366CI.
36. Jovanovic DM, Sterclova M, Mogulkoc N, et al. Comorbidity burden and survival in patients with idiopathic pulmonary fibrosis: the EMPIRE registry study[J]. Respir Res, 2022;23(1):135. DOI:10.1186/s12931-022-02033-6.
37. Cottin V, Selman M, Inoue Y, et al. Syndrome of Combined Pulmonary Fibrosis and Emphysema: An Official ATS/ERS/JRS/ALAT Research Statement[J]. Am J Respir Crit Care Med, 2022;206(4):e7-e41. DOI:10.1164/rccm.202206-1041ST.
本文转载自订阅号「协和呼吸」
原链接戳:间质性肺疾病2022年度综述
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry