
 分享
分享

编前语
80后男性胸闷憋气、活动后加重,CT提示双肺间质性改变,初步诊断为间质性肺炎,但临床多项辅助检查均未能明确间质性肺炎的病因及分型。在诊治陷入僵局时,同在一所医院住在血液科的患者儿子引起了医生们的注意,为什么医生会悄悄的问患者母亲:儿子是亲生的吗?这位患者的诊治为何从儿子的身上找到了突破口?这位80后患者到底得了什么病?
内容来自同济大学附属东方医院举办的第二届「呼吸系统疑难、少见病临床诊治浦江论坛」,现场病例分析可戳文末视频观看。
病例回顾
患者,男性,33岁,入院日期:2014-02-20,主诉:查体发现双肺阴影半年,活动后胸闷憋气3月。
现病史
患者半年前查体行肺CT检查示:双肺慢性炎症,部分呈肺间质纤维化,于当地医院住院治疗,给予应用「左氧氟沙星、头孢西丁」抗感染及「氢化泼尼松(10mg/日)」12天,复查肺CT示肺部阴影较前无明显吸收,出院后未规律治疗。3月前感胸闷憋气,活动后加重,无胸痛,无喘鸣,咳嗽咳痰少,无发热、盗汗,无口干眼干,无关节疼痛等不适,为进一步诊治收入院。
既往史
无高血压、糖尿病史,「汗疱疹」病史多年(未正规诊治),无药物过敏史。
个人史
从事电工工作,曾有建筑工地工作史3年;吸烟史7年,35支/天,10年前已戒;少量饮酒,无特殊药物服用史。
家族史
父亲健在,母亲患有「哮喘」,1姐2妹均体健,否认家族遗传病史。
查体
T:36.8℃,P:94次/分,R:20次/分,BP:115/70mmHg
青年男性,神志清,全身皮肤散在分布色素沉着及色素脱失区域,口唇无紫绀,锁骨上淋巴结未触及肿大,双下肺闻及少许velcro啰音,心率94次/分,律齐,腹软,肝脾未触及,双下肢无水肿,无杵状指(趾)。
辅助检查
胸部CT:双肺胸膜下见斑片状磨玻璃影、模糊小结节灶及纤维网格状影,似乎和我们常见的肺纤维化IPF不太一样,以双上肺为主。右下肺外基底段片状影,边界模糊,局部支气管略显增宽(本院,2013-10-12)。
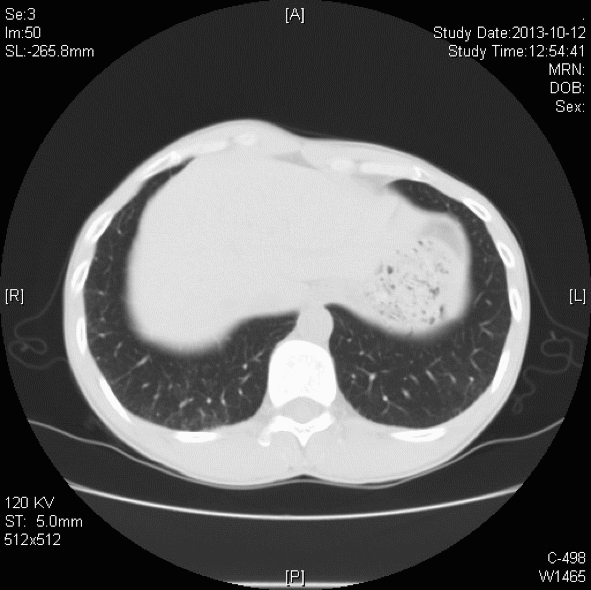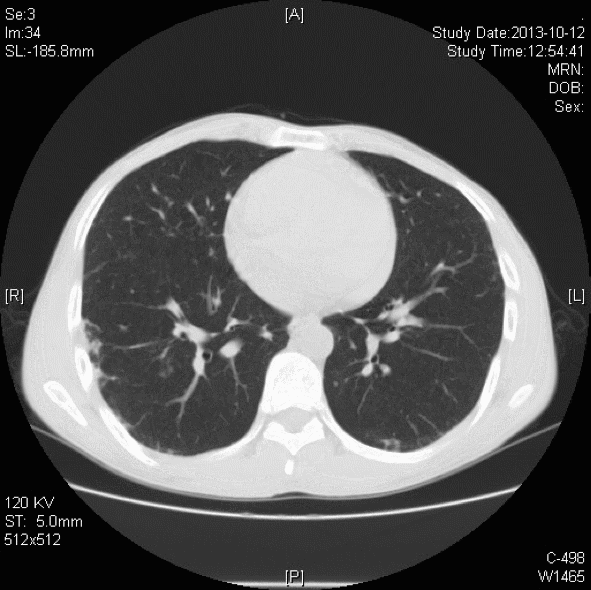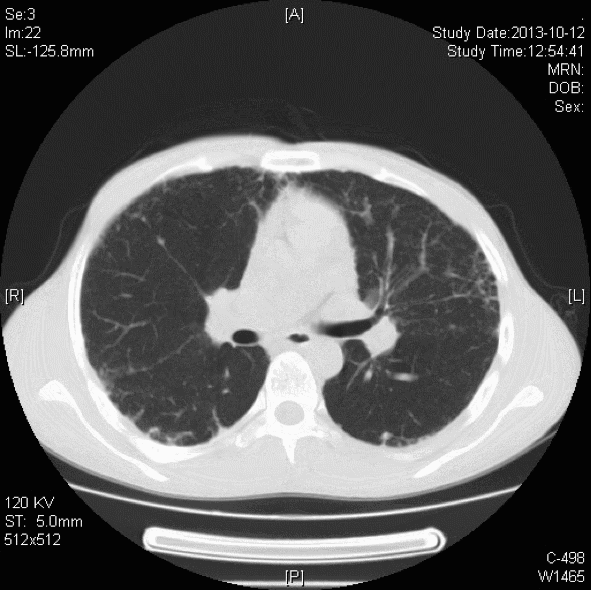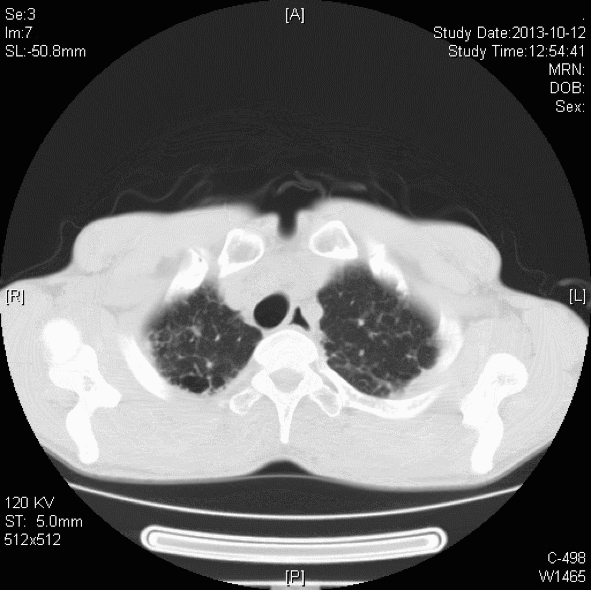
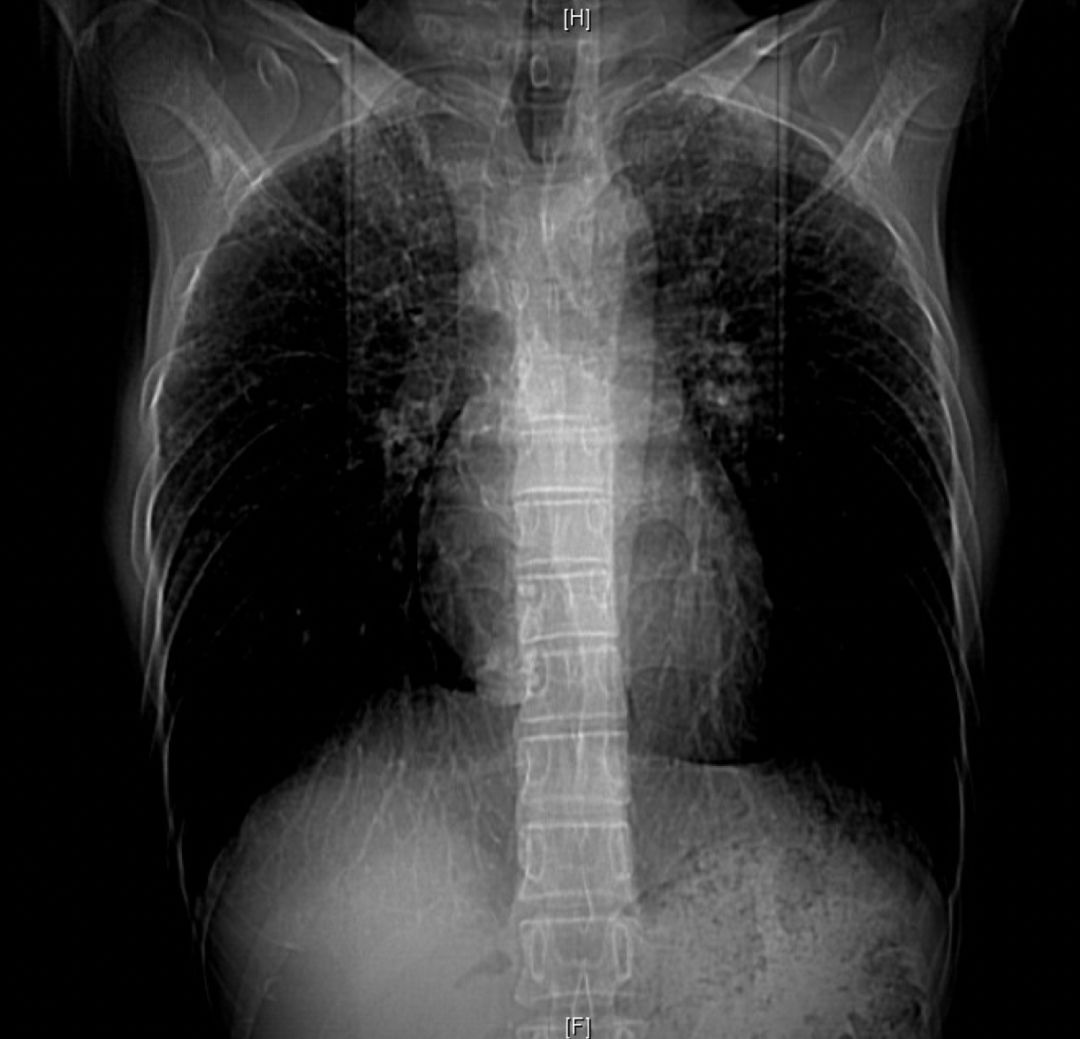
初步诊断
间质性肺炎,汗疱疹?
诊疗经过
辅助检查
血常规:白细胞计数4.31×10^9/L ,血红蛋白127g/L,血小板4.31×10^9/L 。CRP:0.45mg/L(0-5);血凝常规、D二聚体:大致正常。尿常规:阴性。血气分析:PH7.44,氧分压90mmHg,二氧化碳分压46mmHg,氧饱和度95%。
血沉:12mm/;甲功五项:无异常;巨细胞IgM抗体:阴性;真菌G试验:阴性;结核抗体:阴性;肿瘤标志物:无升高;ENA、ANA、ANCA、风湿检测:阴性
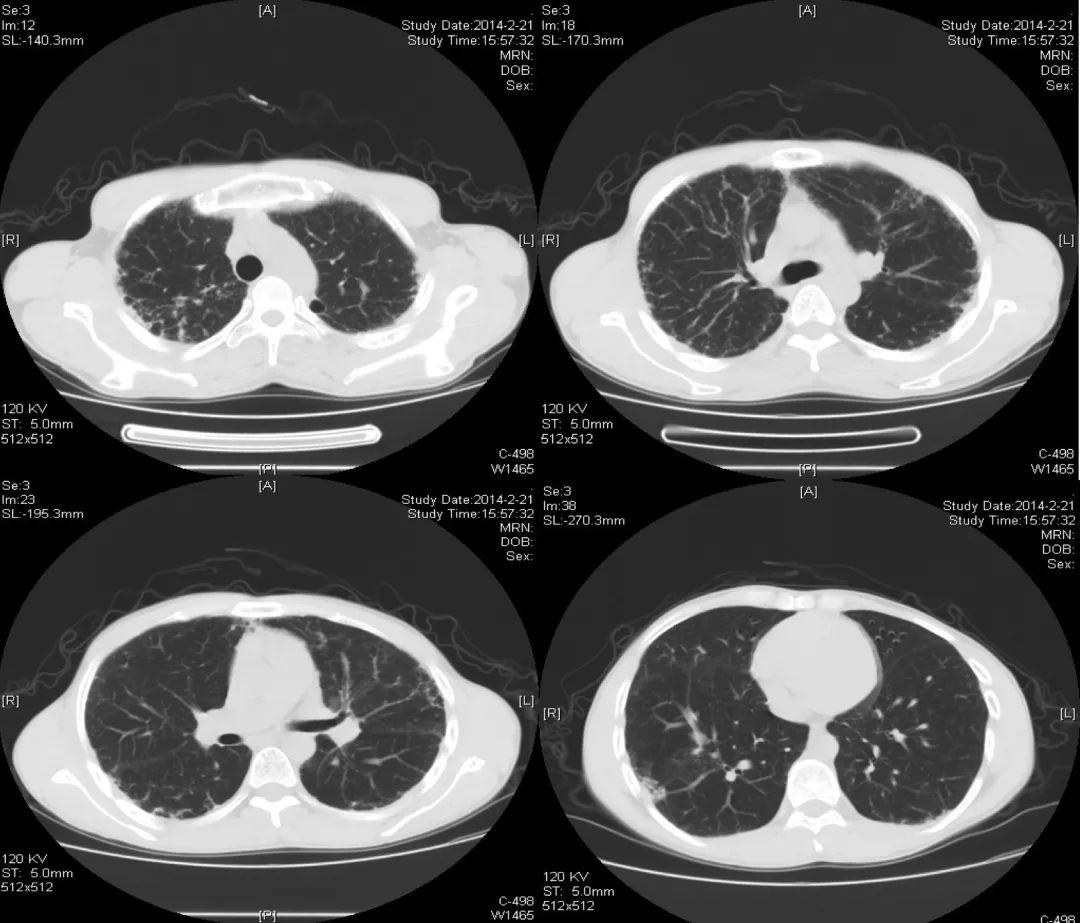
2014-02-21(入院第2天)胸部CT:
发现与2013-10-12胸部CT相似,主要还是以双上肺和胸膜下为主的网格状影,其次右下肺有片状影,可看到充气支气管征。
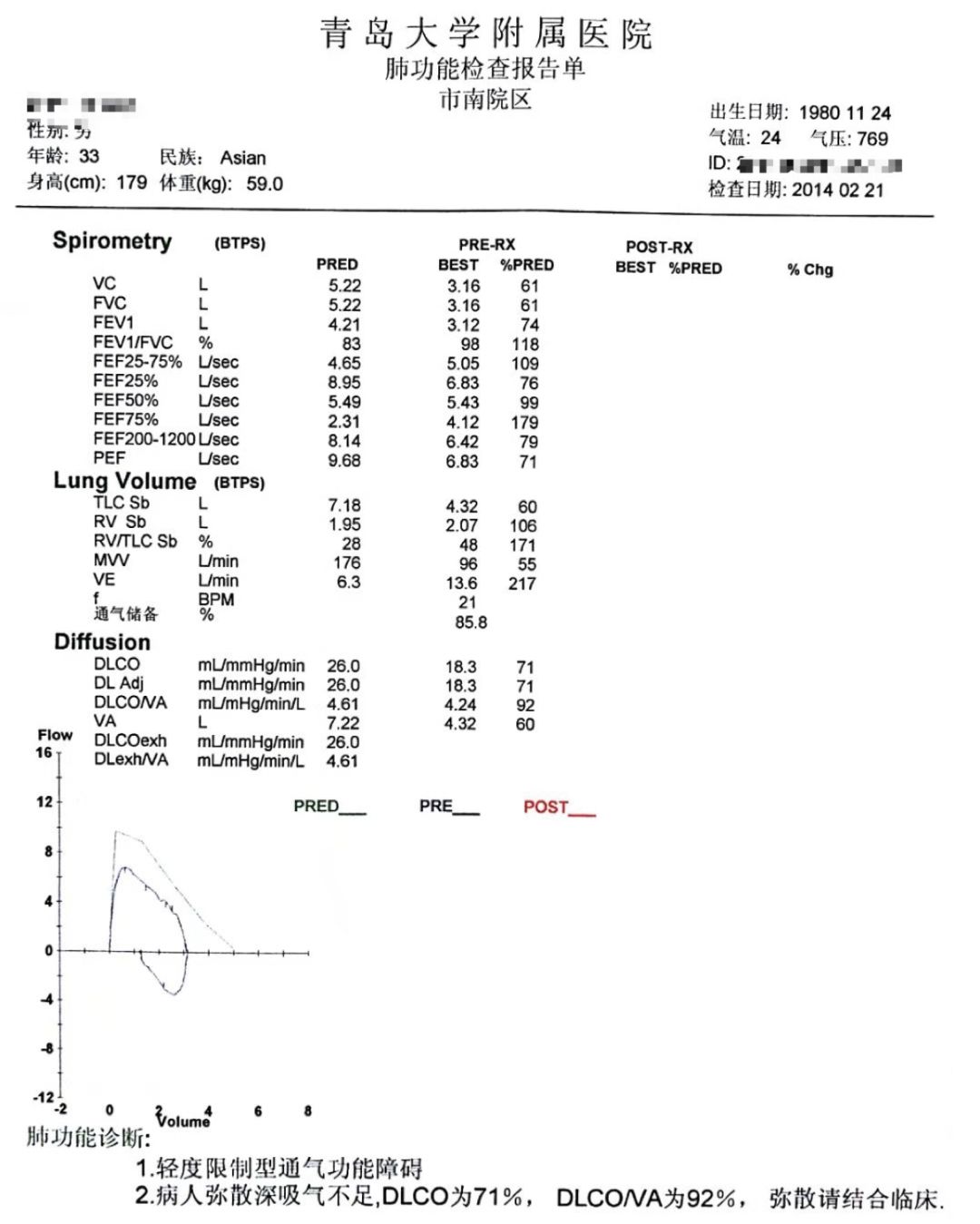
肺功能:轻度限制型通气功能障碍和弥散功能障碍
支气管镜检查:镜下管腔通畅,无异常。

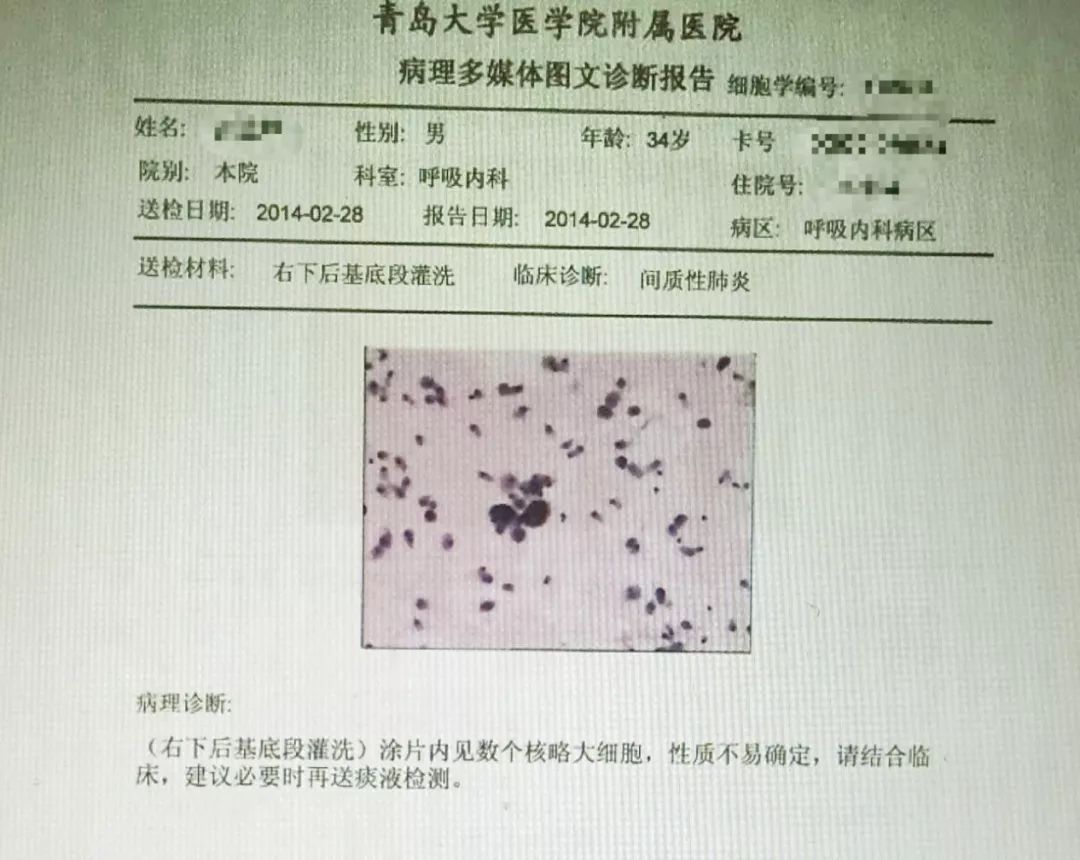
灌洗液病理:(右下后基底段灌洗)涂片内见数个核略大细胞,性质不易确定,请结合临床,建议必要时再送痰液检测。
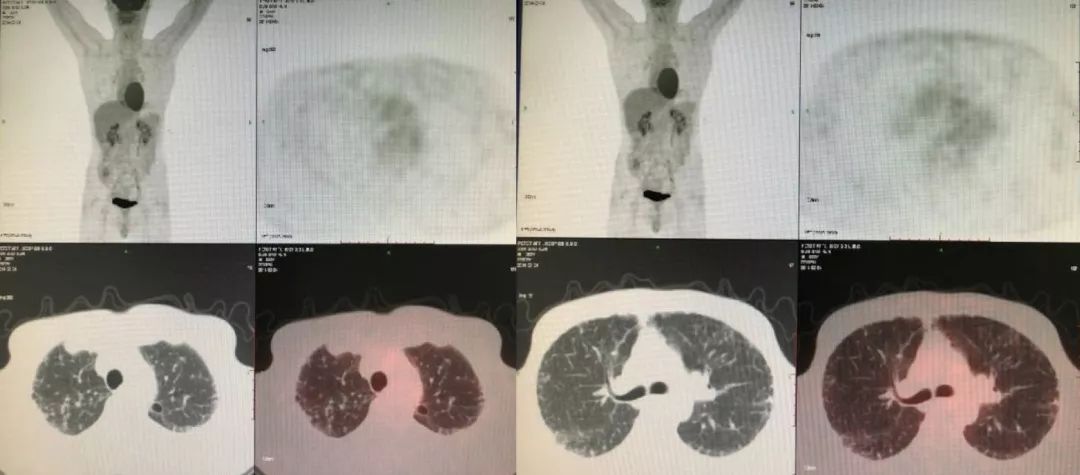
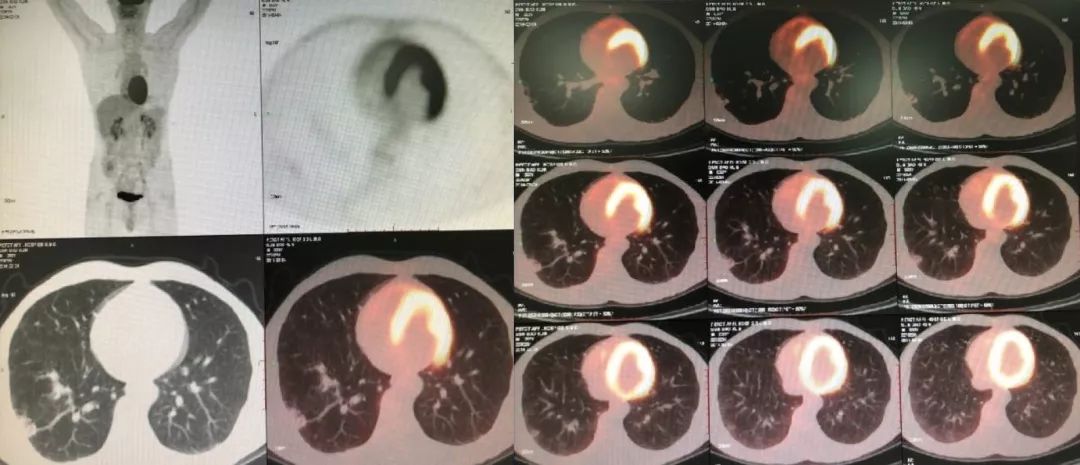
由于右下肺有结节,我们认为不排除肿瘤的可能性,所以做了PET-CT。
PET-CT(2014-03-04):肺间质改变,右下肺结节,SUVmax1.3。SUV小于3.5恶性肿瘤可能性小,故不考虑恶性肿瘤所致间质性肺炎。
做了这么多检查后分析病例特点
> 青年男性,职业电工,既往吸烟史,从事建筑工作3年;
> 发现肺部阴影半年,活动后胸闷憋气3月;
> 影像学示双肺多发网格影;
> 肺功能:限制性通气功能障碍及弥散功能障碍;
> 感染性指标:阴性;
> 血清化验(自身免疫学指标)阴性;
> 气管镜灌洗液病理见数个核略大细胞,性质不易确定;PET-CT无阳性发现;
> 诊断间质性肺炎:病因?分型?
在场嘉宾如何分析诊断疾病?我们有哪些诊疗思路?
▷ 从胸部CT看,右下肺病灶小叶间隔分布,初步印象是淋巴管引流障碍相关性疾病?
▷ 从皮肤的表现来看,有没有可能是全身性的疾病累及到肺?
▷ 目前来看,这个年轻男性从影像学上可以排除一些疾病(如职业为电工,有可能会出现焊工尘肺),应该是遗传性、先天性疾病?
间质性肺疾病的病因及分类
吸入因素:职业(有机/无机)(可能吸入一些电焊气体)、环境(包括家居)、爱好(鸟/宠物)(无)、少量反复误吸
胶原血管疾病、血管炎(无)
肉芽肿疾病:结节病、PLCH(不像)
感染:病毒、PCP、非典型病原体(均无证据)
肿瘤:肺腺癌(肺泡细胞癌)、癌性淋巴管炎?(做过PET-CT;SUV值仅1.3,无证据)
某些特异性疾病:肺泡蛋白沉积症(PAP)、肺淋巴管平滑肌瘤病、慢性嗜酸细胞性肺炎、医源性(药物/放射)(不存在)
继发于其他系统疾病:消化、肾脏、循环等(有皮肤病,其他无)
遗传因素:家族性IPF、结节性硬化、神经纤维瘤病、代谢储存病(无)
特发性间质性肺炎(IIP)(均不符合):主要的IIP:慢性致纤维化性IP(IPF、NSIP)、急性/亚急性IP(COP、AIP)、吸烟相关性IP(DPB、RBILD);少见的IIP:特发性淋巴细胞间质性肺炎、特发性胸膜肺纤维弹性组织增生症;不能分类的IIP:急性纤维素性机化性肺炎、气道中心性间质性肺炎。
以上条件均不符合,无法确诊疾病,我们如何寻找突破口?
1、仔细反复询问病史,既往史、职业史、家族史,对于间质性疾病而言,病史的询问尤为重要
2、认真详细的查体
3、血清化验、胸部影像学、肺功能、支气管镜检查
4、做有创肺活检吗?TBLB、经皮肺活检?胸腔镜肺活检还是开胸肺活检?能给我们提示吗?
5、反复阅读,仔细推敲患者辅助检验检查结果
6、查阅文献
果然发现了一些线索
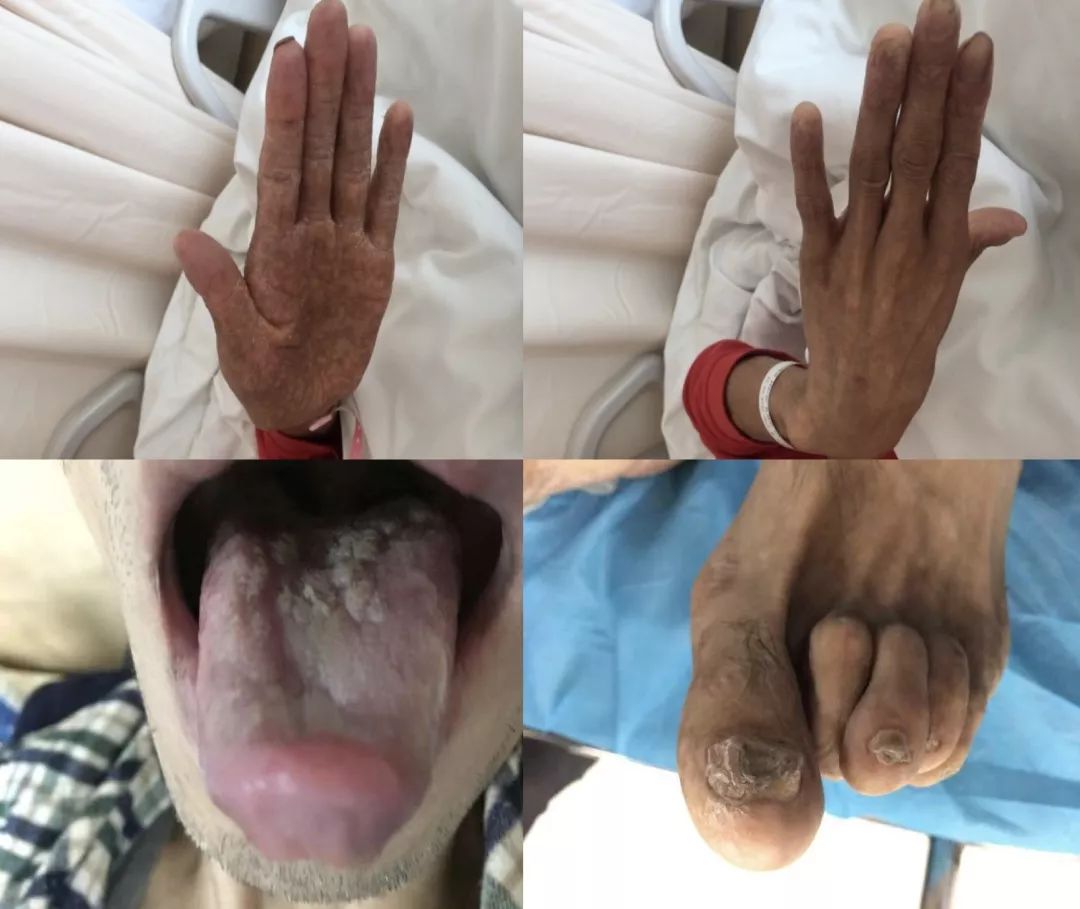
1、须发灰白、指甲薄如蝉翼
查体:患者仅33岁,头发、胡须就都是灰白的;全身皮肤改变(手指比较细长;手心和手背皮肤有改变,主要为色素脱失和色素沉着交替存在,部分有脱屑);粘膜改变(口腔粘膜改变、舌体白斑);指(趾)甲改变(脚趾指甲变形,手指指甲有开裂、变薄、缺失);
诊疗经过
皮肤科第1次会诊:PE:患者全身皮肤大理石样改变,无自觉症状,双手掌部可见皮肤粗糙,干燥脱屑。考虑:1、皮肤异色症(全身皮肤)2、湿疹(掌部)可能;建议真菌镜检。
辅助检查:真菌镜检:

发现菌丝阳性,于是皮肤科第2次会诊,会诊意见:患者双手部皮肤粗糙干燥脱屑,真菌镜检(+)。考虑「手藓」可能,建议必亮外用Bid。
2、儿子患再生障碍性贫血
病史:
既往史:幼时(8岁起)指甲变薄,开裂,脱落,眼睛频繁流泪;
职业史:电工,建筑工地工作史3年;
家族史:儿子患再生障碍性贫血,目前反复在我院儿童血液科治疗。
这里思路可能就清楚了许多,我们再次进行了临床诊断分析
临床表现:①肺纤维化(主要);②青年男性:灰白头发及胡须;③皮肤粘膜改变:全身皮肤大理石样改变,色素沉着与脱失交替分布,掌心皮肤干燥脱屑;口腔粘膜白斑;④指甲改变 :指(趾)甲发育不良,萎缩变薄,变尖,变细,无指甲;⑤流泪:鼻泪管阻塞。
家族史:儿子患再生障碍性贫血。
那么患者的肺纤维化和他儿子的再生障碍性贫血有什么关系?是不是遗传性疾病?基因检测有必要吗?
我们顺藤摸瓜,正好患者儿子在我院儿童血液科住院,翻看孩子的病例,这个患者的诊断一下清晰明了!
患者儿子简要病史
患者儿子目前12岁,4岁时诊为「再生障碍性贫血」,常规治疗效果不佳,9岁时就诊于中国医学科学院血液病医院,行骨髓细胞学及骨髓病理学检查,基因检测示:TINF2基因的一个杂合变异(c.849:缺失G,来源于父亲),符合常染色体显性先天性角化不全症3型。诊断为「先天性角化不全」,10岁时在北京某医院行异基因造血干细胞移植,此后反复在我院儿童血液科治疗。
最终诊断
先天性角化不全症
随访:活动性呼吸困难逐渐加重,出现了气胸
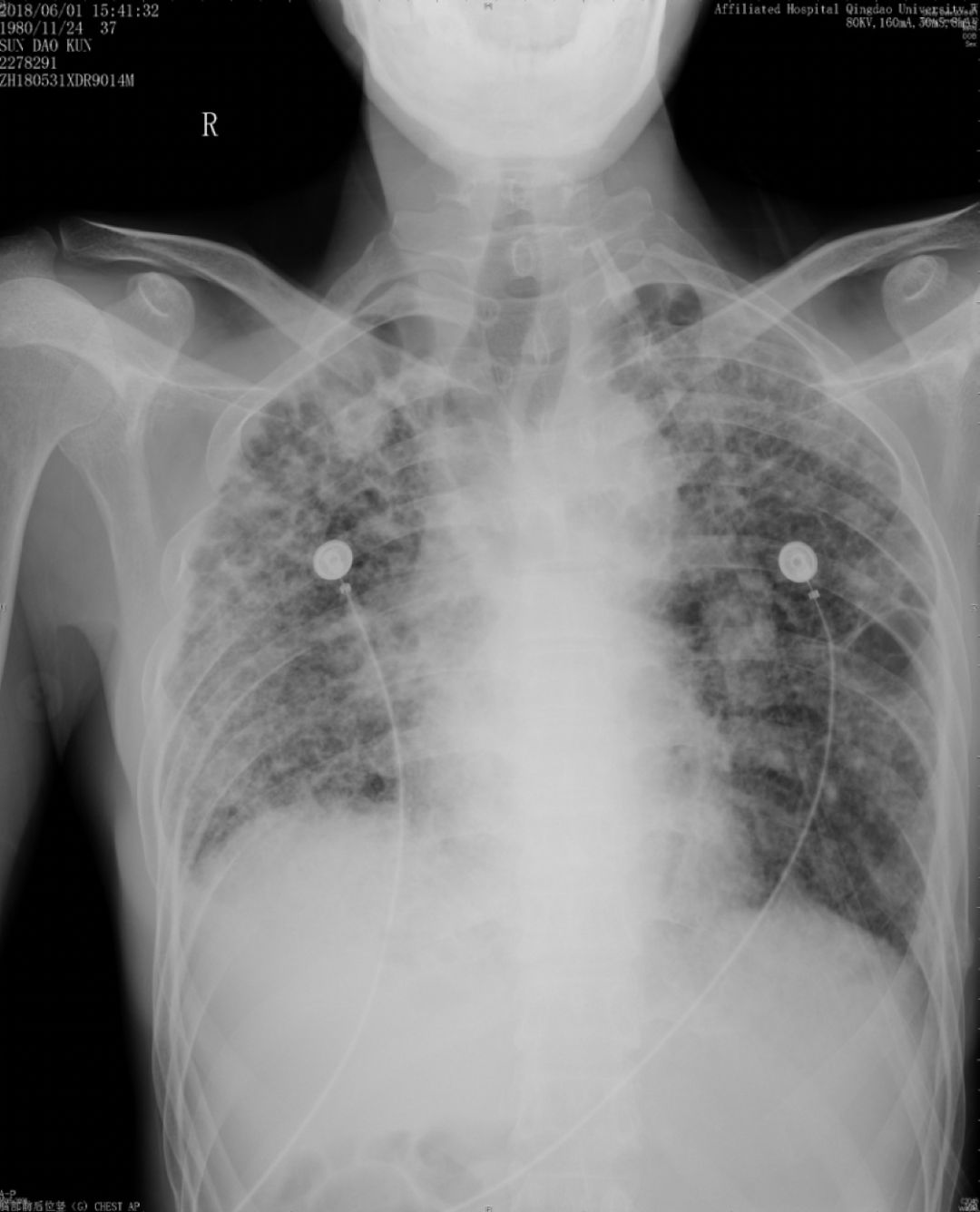
由于儿子反复治疗的花销已经很大了,这位患者诊断后没有特别治疗,曾去过多家医院,考虑过肺移植,但由于是遗传性疾病只好作罢。随后在我院随访,呼吸困难逐渐加重,间质改变越来越严重,原来是上肺比较明显,后来下肺病变也加重了,再后来右上肺出现大泡,最后出现气胸,反复住院,这个患者后来再也没有来了……
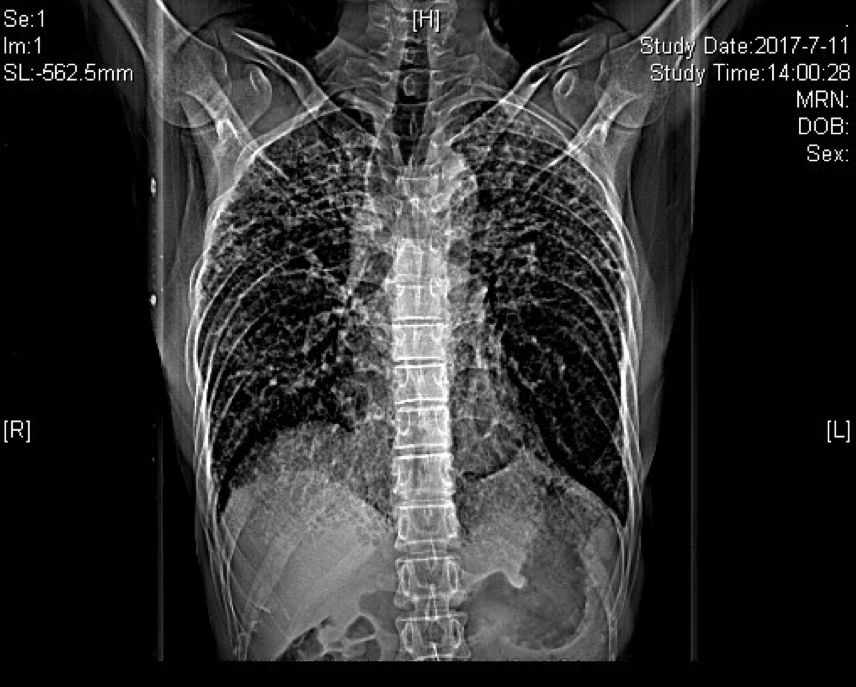
胸部CT(2017-07-11):
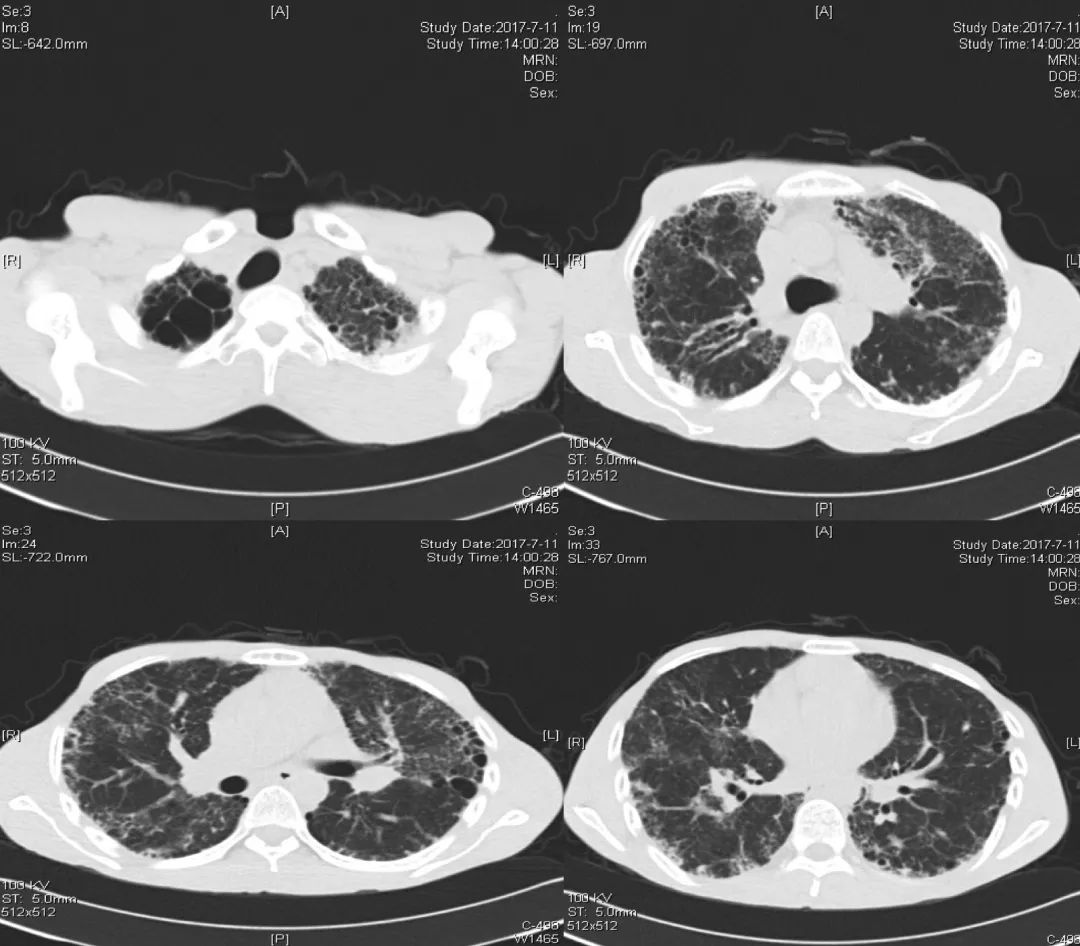
胸部CT(2018-02-22):
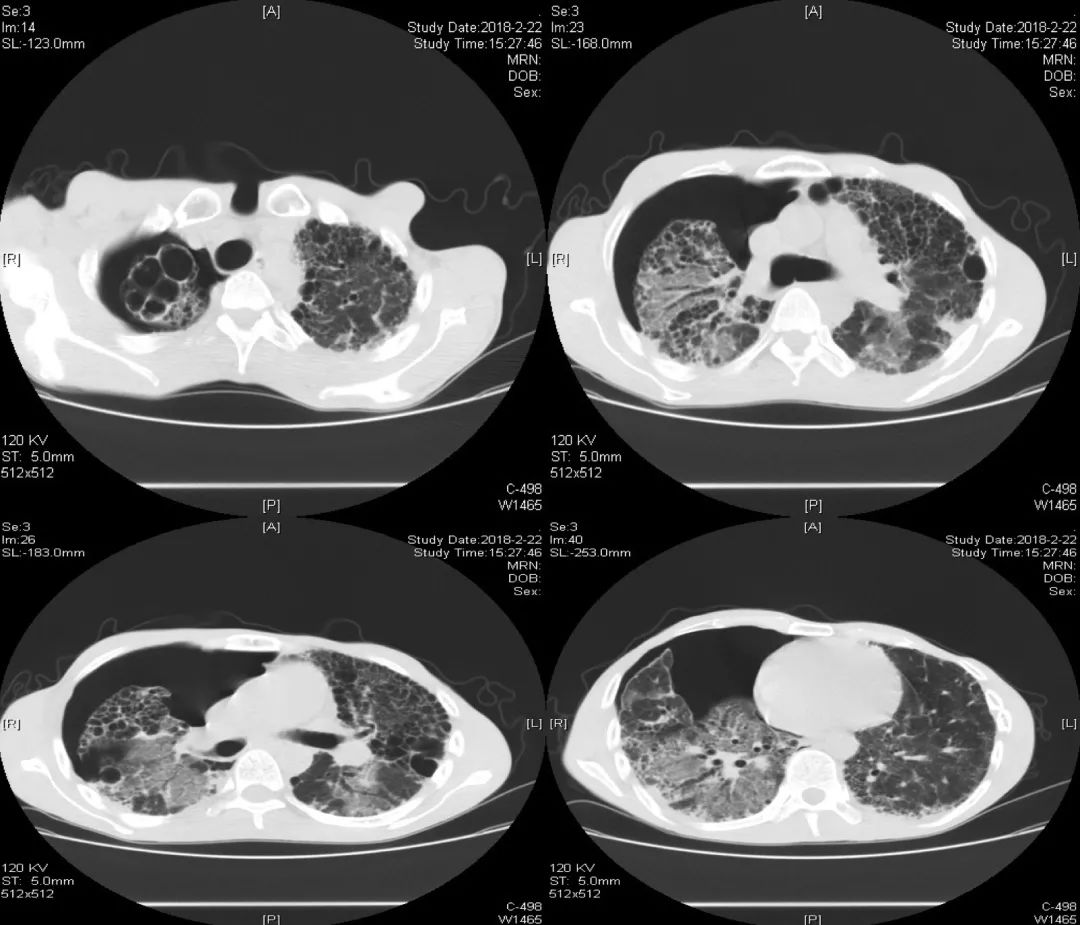
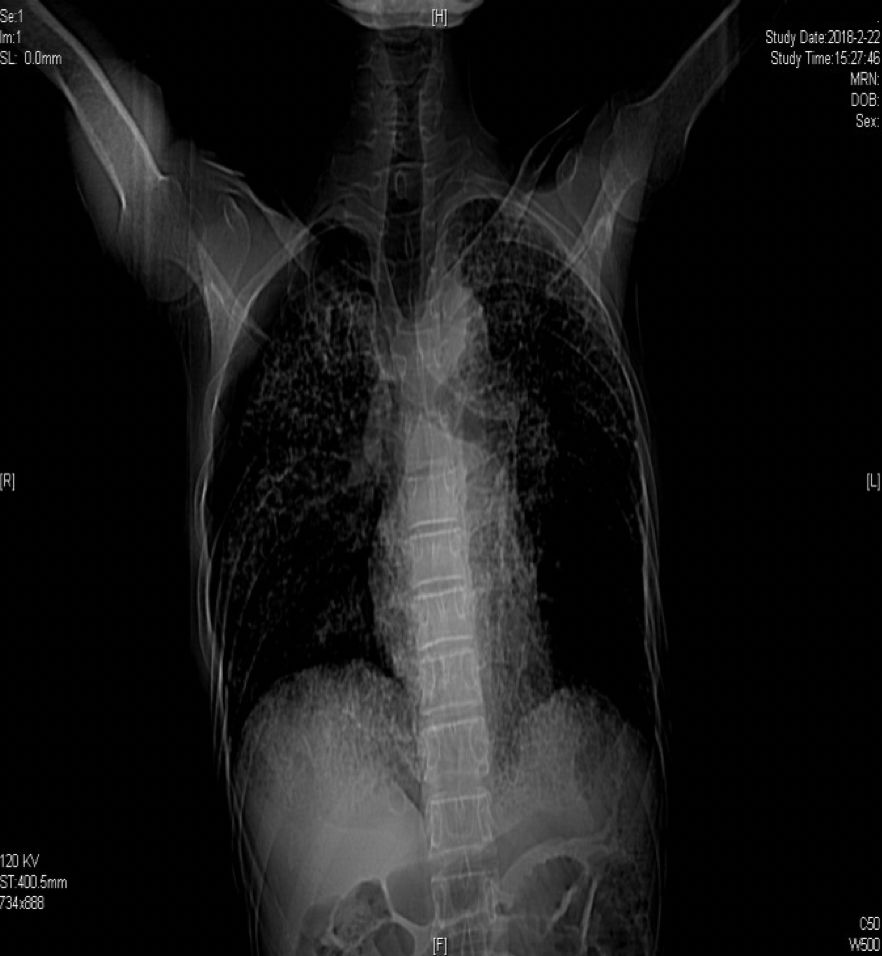
胸部正位片(2018-03-01):
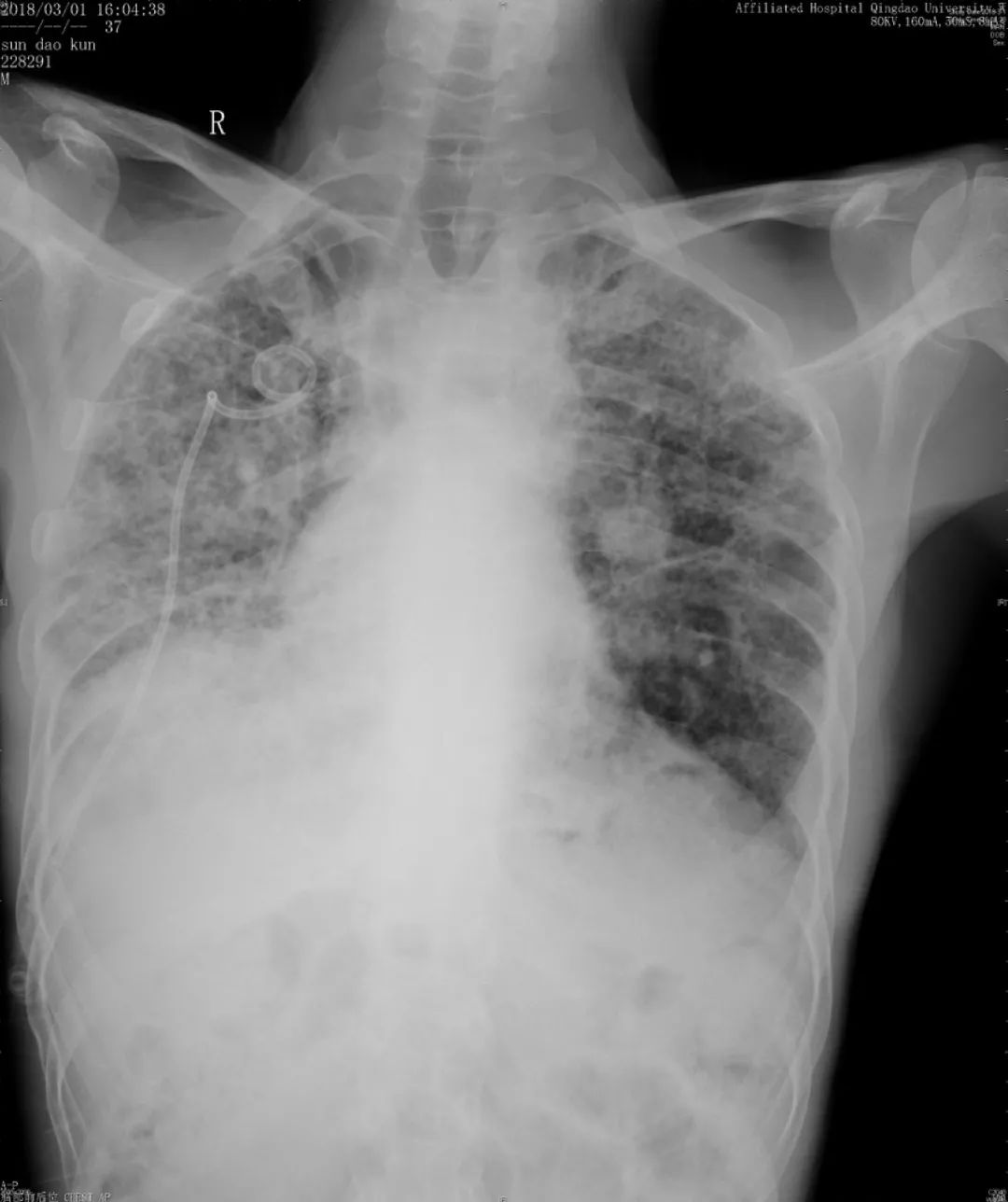
胸部正位片(2018-06-01):最后一次
了解先天性角化不全症这个疾病
1、概述
先天性角化不全症(简称DKC)也叫做Zinsser-Engman-Cole症候群,是一种罕见的进行性骨髓失能症候群。
这个疾病主要有三种特征:
① 网状皮肤色素沉着过多;
② 指(趾)甲营养失调;
③ 口腔黏膜有白斑。
于此疾病中可发现染色体端粒酶功能不良、核糖体缺乏和蛋白质合成功能异常等现象。早期死亡的原因常与骨髓失能、感染、致死的肺部并发症,或恶性肿瘤有关。
2、遗传模式
目前发现,导致DKC的各种端粒酶相关基因突变,主要包括DKC1、TERC、TERT、NOPl0、NHP2、POTlb和TINF2等。近年国外较大样本数据显示,上述基因突变约涵盖60%的患者,存在X染色体隐性、常染色体显性和常染色体隐性等3种遗传模式,另有约40%的致病基因尚未明确。
3、症状特征
先天性角化不全症患者通常在10岁左右,会开始出现典型症状,常以皮肤色素沉着和指(趾)甲异常等先表现。表皮黏膜的典型的特征,通常在5~15岁时出现。皮肤网状色素沉着过多、指(趾)甲营养失调和黏膜白斑症为先天性角化不全症三个主要的典型症状。
4、临床表现
1、皮肤:主要是皮肤不正常色素沉着,可见杂色班纹状或是网状的黑到灰色的暗色斑块或是浅色色斑。主要发生在上部躯干、颈部和脸。其他症状,包括头皮、眼眉和睫毛的脱发症;过早灰白发;掌心皮肤角化过度。
2、指(趾)甲:有90%病患会有指(趾)甲营养失调的情形,通常手指先发生。指(趾)甲开始先隆起而后纵向裂开,并持续萎缩、变薄和翼状化,最后导致指甲变小或消失。
3、黏膜:约80%患者会有典型黏膜白斑症,通常发生在口腔黏膜、舌头和口咽部。其它黏膜,如食道、尿道开口、龟头、泪管、结膜、阴道、肛门等,也可能受影响。这些区域发生收缩和狭窄,最后导致吞咽困难、排尿困难、包茎和泪溢。
4、骨髓失能:约有90%的个案会有外周血细胞减少情形。有些病患会以此为最初表现的症状,通常在10岁左右开始出现。骨髓失能是造成死亡的重要原因,约有70%是因为骨髓失能而造成出血和机会性感染。
5、肺部合并症:约有20%先天性角化不全症的个案会有肺部合并症,肺部纤维化和肺部血管系统的异常。患者应避免服用会产生肺部毒性的药物;并且在骨髓移植期间,保护好肺部。
6、增加恶性肿瘤的风险:患者较高的恶性黏膜瘤发生率,特别是鳞状上皮细胞癌,如口腔、鼻咽、食道、直肠、阴道或者子宫颈。这些地方经常发生黏膜白斑病。皮肤的鳞状上皮癌的流行率也会升高。其他恶性肿瘤包括何杰金氏淋巴瘤、胃肠道的腺癌和支气管和喉头癌。通常在30岁左右开始出现恶性肿瘤。
7、神经系统:患者会有学习困难和智力缺陷的问题。
8、眼睛:有80%先天性角化不全症患者会有结膜炎、眼睑炎及泪管狭窄导致的溢泪。
9、骨骼系统:患者可能会有下颔骨发育不全、骨质疏松、无血管的坏死和脊柱侧弯。
10、胃肠系统:包括食管曲张、肝脾肿大和肝硬化。
11、生殖泌尿系统:Hypospastic睾丸、尿道下裂和输尿管狭窄。
12、女性带因者:先天性角化不全症的女性带因者也许有少数的临床症状。一项最近研究表示,20位女性带因者中有三位有临床症状,包括:单一营养不良的指(趾)甲、浅色色素斑、或轻微黏膜白斑病等临床症状。
5、诊断
1、典型表现(三联征):①皮肤色素沉着:弥散性棕褐色或灰褐色、斑点状或网格状的色素沉着,可融合成片。常始发于面部和颈胸部,可逐渐扩展至全身。②黏膜白斑:舌面和颊黏膜增厚性白斑。③指(趾)甲发育不良:指(趾)甲逐渐生长缓慢、增厚变形或萎缩,直至最终脱落。
2、皮肤活检:表现为角化过度或角化不全,表皮萎缩;真皮层黑素颗粒沉着及噬黑素细胞聚集。
3、相关基因检测:端粒酶相关基因检测。
6、治疗
血细胞下降是DKC的严重表现和主要致死原因,故为目前国内外药物治疗研究重点。
① 小剂量雄性激素可促进端粒酶活性,有效率可达60~70%;
② 小剂量糖皮质激素,可联合雄性激素,有效率约70%左右;
③ allo-HSCT,移植成功可获得造血功能重建;
④ 外源性TERC疗法,实验研究发现采用外源性TERC治疗DKC1和TERC突变者,有助于恢复端粒酶活性和端粒长度,但其可行性有待临床;
⑤ 因为DKC与获得性再障的免疫介导发病机制完全不同,所以不能采用IST治疗。
7、预后
先天性角化不全症为一多重复杂的疾病其预后不佳,平均存活年龄为30岁。大部分的死因与感染、出血和恶性肿瘤有关。有纪录的先天性角化不全症约有70%个案死亡是因为骨髓失能或其合并症所致,这些个案死亡的平均年龄为16岁。
从这个病例中我们发现了临床中还有的问题
1、遗传性疾病:患者儿子基因检测结果明确,为常染色体显性先天性角化不全症,那这个患者的遗传方式是什么?他的父母及兄弟姐妹身体都没有特殊疾病,而且还私下询问了患者母亲,确证是具有血缘关系的母子。既然是常染色体显性遗传,为什么患者的父母、姐妹都未患病?患者有2个孩子,儿子患病,另一个4岁的女儿目前状况良好,那以后会不会发病?
2、家系图的绘制:血样采集
3、治疗:再生障碍性贫血可以进行干细胞移植,肺纤维化可不可以肺移植呢?移植后还会不会再发病?
心得体会
间质性肺疾病是一组异源性疾病,病因众多,临床表现各异,诊断和治疗均存在困难。在分析具体原因时,注意影像学形态、分布,需要把常见的病因逐一考虑、排除;对于找不到原因者,还需要考虑少见情况,不要轻易下「特异性」定义。询问病史、查体也很重要,这样才能「山重水复疑无路,柳岸花明又一村」。
专家介绍

王红梅
教授,博士,硕士研究生导师;青岛大学附属医院黄岛院区呼吸内科主任,中华医学会山东呼吸分会青年委员,山东医师学会呼吸分会委员肺癌协作组副组长,中国呼吸肿瘤协作组成员。

郭彩宏
青岛大学附属医院呼吸内科主治医师;山东省抗癌协会肿瘤微创治疗分会委员;山东省老年生物靶向治疗专业委员会委员;山东省康复医学会呼吸康复专业委员会委员;主要专业方向为呼吸疑难少见病的诊治,肺部肿瘤的基础与临床。

任敦强
青岛大学附属医院呼吸与危重症医学科主治医师,医学博士,中国中西医结合学会变态反应专业委员会哮喘学组委员,毕业于广州医科大学/广州呼吸疾病研究所,博士期间师从钟南山院士,主要研究方向:间质性肺疾病、慢性气道疾病发病机制及药物靶点筛选。
*特别感谢王红梅教授给予的指导和帮助
来源:第二届「呼吸系统疑难、少见病临床诊治浦江论坛」
点击下方视频,看郭彩宏医生讲解不同寻常的肺纤维化:
建议在Wi-Fi环境下观看