
 分享
分享
推荐语
近日,国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布了《第一批罕见病目录》,共收录121种疾病,其中多种为呼吸疾病,为此《呼吸界》特约专家对此逐一进行分析解读,敬请关注。
囊性纤维化(cystic fibrosis,CF),也被称为囊性纤维变性。是由囊性纤维化转膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)基因突变导致的多系统疾病。是高加索人种最常见的常染色体隐性遗传疾病之一。
不同国家和地区报道的发病率不一,新生儿发病率约1/1800~25000。亚洲和非洲发病人数远远少于欧洲和北美洲,CF在我国尚处于初步认识阶段,其确切的发病率尚不清楚。
临床表现
CF患者常在婴幼儿或青少年期起病,男女比例大致相等。肺脏是最常见的受累器官,表现为反复咳嗽、咳痰、咯血、发热,影像学表现为上肺为主的弥漫性支气管扩张(图1)。病原学检查显示慢性细菌感染及病原体的定植,并最终出现铜绿假单胞菌或洋葱克雷伯杆菌等慢性感染。
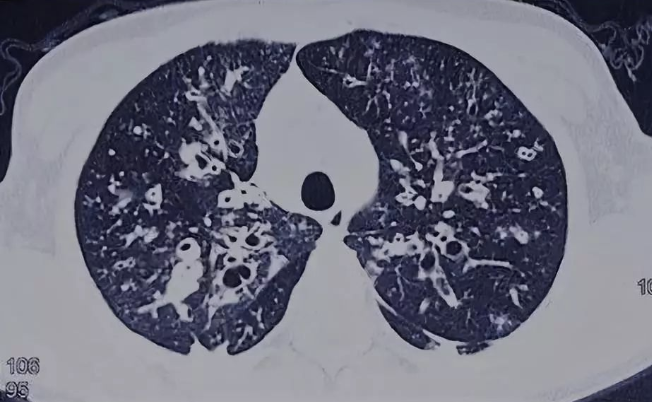
图1
消化道方面,粘稠的胆汁和胰液的流动异常可以导致消化或吸收不良,或是肝脏、胰腺疾病,部分患者可合并糖尿病。CF患者还会出现肠梗阻和直肠脱垂。男性患者可表现为先天性双侧输精管缺失(CBAVD)。
病因和发病机制
CF为常染色体隐性遗传(AR),其致病基因CFTR位于7q,目前已发现逾2000种CFTR基因突变(CFGAC,www.genet.sickkids.on.ca),其表达的CFTR蛋白含1480个氨基酸残基,是上皮细胞表面的一种氯离子通道蛋白,当CFTR发生基因突变时,CFTR蛋白所在的上皮细胞分泌氯离子和水分减少,钠离子重吸收增加,导致细胞内高渗环境及分泌物粘稠,从而造成管腔阻塞而致病。欧美最常见的的突变是△F508,但是在东亚CF患者中(中国,韩国,日本,越南,泰国)极为少见。目前研究认为中国人CF最常见的突变为G970D。
诊断
至少一个器官存在CF的典型表现以及存在以下至少一种CFTR基因功能异常的证据:
(1) 2个部位汗液氯离子测定超过60mmol/L;
(2) 等位基因上存在2个CFTR致病突变;
(3) 鼻电位差异常。
鉴别诊断
需要与可以导致弥漫性支气管扩张的疾病相鉴别。这些疾病包括:感染后支气管扩张,弥漫性泛细支气管炎,α-1抗胰蛋白酶缺乏(AAT),原发性纤毛运动障碍(PCD),变应性支气管肺曲霉菌病(ABPA),低丙种球蛋白血症,胃食管反流病等。
治疗 目前仍没有治愈的方法
CF是一类慢性的、终生性疾病,在不同的年龄阶段需要不同的治疗,但目前仍然没有治愈的方法。常见的治疗建议包括:
(1)促进气道分泌物的清除:吸入DNase Ⅰ(α-链道酶),高渗盐水,N-乙酰半胱氨酸,体位引流和叩击形式的胸部理疗等。
(2)抗生素治疗:反复出现感染的CF患者采用针对铜绿假单胞菌的雾化抗生素(妥布霉素和氨曲南等)长期治疗。
(3)支气管扩张剂:缓解呼吸困难。
(4)抗炎治疗:大环内酯类药物可改善CF患者的呼吸功能并减少肺部疾病加重的发生率。CF基金会指南委员会建议给6~17岁肺功能良好(即FEV1>预计值的60%)的儿童使用大剂量布洛芬。但是如果患儿超过13岁,不再推荐初始给予布洛芬治疗。
(5)改善营养:CF患者如存在消化和吸收的障碍,可以补充胰酶和微量元素等。
(6)肺移植:终末期CF肺病患者的治疗选择。[4,5]
随着对CFTR基因的深入研究,治疗开始针对解决CFTR基因缺陷的源头问题。CFTR调节剂通过改善缺陷的CFTR蛋白功能而发挥作用。代表性药物为依伐卡托(Ivacaftor),它能够修复突变CF蛋白的功能,其作用的程度和广度都显著超过了当前可用于CF的任何其他疗法。依法卡托适用的突变靶点包括G178R、S549N、S549R、G551S、G1244E、S1251N、S1255P、G1349D或R117H。对于ΔF508纯合突变的患者,鲁玛卡托(lumicaftor)联合依伐卡托治疗能够轻度改善肺功能,降低肺部疾病加重的风险。目前针对中国人基因突变的靶向治疗还未有研究。
预后 患者的预期寿命已超过40岁
随着对疾病认识的深入和治疗方法的进步,欧美国家报道CF患者的预期寿命已超过40岁。随着基因精准治疗的进展,相信在不远的将来,CF患者的预后将得到更大程度的改善。
患者的兄弟姐妹也可能同样患病?遗传咨询与产前诊断怎么做?
CF以AR方式遗传,因此,每个CF患者的父母怀孕生育下一胎时都有25%的可能性为患者;50%的可能性为无症状的携带者;25%的可能性为完全正常。如果家系中先证者的两个CFTR致病突变均明确,即可给家庭提供产前诊断或胚胎植入前基因诊断。
由于中国人的基因与欧美常见基因不同,如果临床发现高度可疑的CF患者,常用的基因筛查手段并无阳性发现时,大片段缺失的筛查是必要的。
参考文献
[1] Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med, 2005,352(19):1992-2001.
[2] Singh M, Rebordosa C, Bernholz J, et al. Epidemiology and genetics of cystic fibrosis in Asia: In preparation for the next-generation treatments. Respirology,2015,20(8):1172-1181.
[3] Tian X, Liu Y, Yang J, et al. p.G970D is the most frequent CFTR mutation in Chinese patients with cystic fibrosis. Hum Genome Var, 2016, 3: 15063.
[4] Mogayzel PJ, Jr., Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med, 2013,187(7):680-689.
[5] Mogayzel PJ, Jr., Naureckas ET, Robinson KA, et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc, 2014,11(10):1640-1650.
作者介绍
