
 分享
分享
摘要
本文报道1例以反复肺出血为表现的血管型Ehlers-Danlos综合征。患者男,22岁,因「间断咯血、胸痛18个月」入院。胸部CT示双肺多发结节及空洞,周围伴晕征。血清炎症指标包括血沉、C反应蛋白、白介素-6均在正常范围,经多次支气管镜、肺活检病原及病理学检查未能明确诊断,曾给予经验性抗细菌、抗真菌、抗结核、抗寄生虫治疗,肺内病变仍表现出此消彼长的特点。胸腔镜肺活检病理见肺出血、血肿、骨化及纤维性结节,提示血管型Ehlers-Danlos综合征的可能性。最终经基因检测发现COL3A1突变,诊断为血管型Ehlers-Danlos综合征。该患者无皮肤松弛、关节过伸等肺外表现。随诊3年,患者除间断有少量咯血外,尚未出现其他血管或脏器受累证据。对于以反复咯血为表现,肺内表现为此消彼长的结节、空洞或囊腔而无明显炎症反应、对抗感染治疗无效的年轻患者,应考虑到血管型Ehlers-Danlos综合征的可能性,尽早进行基因检测,避免不必要的有创检查对患者的伤害。
患者男,22岁,因「间断咯血、胸痛18个月」于2019年3月25日入院。患者2017年9月起无明显诱因出现间断咯少量鲜血,伴胸部隐痛,无痰,无发热。胸部CT示双肺多发大小不一结节影,伴晕征,部分结节伴空洞,双肺散在片状磨玻璃影,无纵隔淋巴结肿大(图1)。先后给予静脉头孢克肟联合阿奇霉素(具体剂量不详)抗感染1周、口服左氧氟沙星联合法罗培南抗感染1个月,肺内结节空洞部分吸收(图1,2),部分新发。

图1 胸部CT(2017年9月12日)示左上叶结节空洞伴周围磨玻璃密度影
图2 胸部CT(2017年9月25日)示左上叶结节缩小,空洞消失,周围磨玻璃密度影吸收
先后就诊于多家医院,查血常规、ESR、C反应蛋白均正常,淋巴细胞干扰素释放试验、隐球菌抗原、1-3-β-D-葡聚糖、曲霉半乳甘露聚糖、寄生虫抗体均阴性,抗核抗体谱、抗中性粒细胞胞质抗体(ANCA)、抗肾小球基底膜抗体均阴性。肺功能、超声心动图均正常。先后3次行支气管镜检查,支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)未检出致病菌。经皮肺穿刺活检提示肺泡腔内见含铁血黄素细胞,未见肿瘤细胞。先后试验性使用抗真菌(氟康唑0.6 g,1次/d,治疗2周,伊曲康唑0.2 g,1次/d,治疗1个月)、抗结核(HRZE四联治疗3个月)、抗寄生虫(吡喹酮1.6g,3次/d,治疗3 d,2个疗程)。治疗期间及治疗间期,患者仍有间断少量咯血及胸痛,曾出现左侧气胸,后自行吸收(图3,4)。
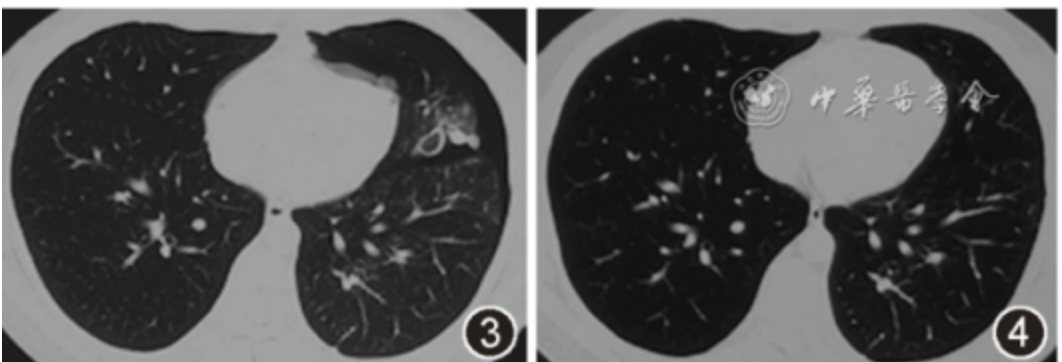
图3 胸部CT(2018年3月28日)示左上叶下舌段小囊状病变及多发实性结节,周围少量磨玻璃密度影,左侧少量气胸
图4 胸部CT(2018年4月29日)示原左上叶下舌段囊状病变及结节均消失,左侧气胸完全吸收
定期复查胸部CT,肺内病灶消退及新发,双肺条索影逐渐增多。2018年11月复查胸部CT:双肺多发结节伴周围磨玻璃影,部分较大结节内部见气液平,左下叶胸膜下局灶肺气肿。病程中无发热,无其他脏器出血。
既往史:患者自幼易出现口腔溃疡,平均每月1次,无生殖器溃疡、皮疹、眼部不适。无疫区旅居史或疫水接触史,不吸烟。无家族遗传病史。入院体检:生命体征平稳,身高172 cm,体重80 kg。浅表淋巴结无肿大,双肺未闻及干湿性啰音,心腹体检无异常,皮肤关节体检无异常。
讨论
张婷(呼吸与危重症医学科):
患者为青年男性,慢性病程。反复出现咯血及胸痛症状。肺内反复出现结节、空洞、囊状影伴周围磨玻璃影,并出现过自发性气胸。患者在1年半的时间内经历了12次胸部CT、4次支气管镜、3次经皮肺穿刺活检,未能得到明确诊断。先后使用抗细菌、抗真菌、抗结核及抗寄生虫药物,均无明确疗效。肺部结节空洞呈现此消彼长,自愈与复发交替的游走性特点。
彭敏(呼吸与危重症医学科):
对于游走性肺内结节的鉴别诊断包括:(1)肺血管炎:结合病变部位,倾向于中小血管受累。①ANCA相关性血管炎:肉芽肿性多血管炎可出现肺内结节空洞自行消退的特点,但患者无鼻窦、肾脏等肺外器官受累表现,炎症指标正常,ANCA阴性,不符合诊断。②白塞病:肺内病变以肺动脉瘤伴或不伴出血为主,亦可出现肺栓塞或肺梗死,但出现空洞者少见。患者虽有复发性口腔溃疡,但无生殖器溃疡、葡萄膜炎等其他典型表现,尚不能诊断。③其他不能分类的肺血管炎:少见情况下,孤立性肺血管炎可能无肺外脏器受累,难以归入现有分类标准,诊断依赖病理,且多需外科肺活检,目前不能排除。(2)感染性病变:细菌、真菌、分枝杆菌等病原体引起的肺部感染,可出现治疗有效,但停药后复发的情况。复发通常在停药一段时间后出现,与本患者肺内病灶消失与新发同期出现的特点不符。寄生虫感染时,因虫体有移动性,可表现出游走性特点,但多伴有外周血嗜酸细胞及总免疫球蛋白E升高。本患者外周血嗜酸细胞及总免疫球蛋白E始终正常,且寄生虫抗体阴性,抗寄生虫治疗无效,不支持寄生虫感染。(3)机化性肺炎:可出现游走性特点,但一般不出现肺空洞、出血,肺活检病理亦不支持,可排除。(4)女性患者还需考虑子宫内膜异位症,但本例为男性患者,可排除。综上,患者病变与血管关系密切,但上述常见疾病均暂不符合诊断标准,建议行外科肺活检。
高莉(医学影像科):
患者胸部CT有以下特点:(1)肺内多发结节周围伴磨玻璃影,结合咯血症状,提示肺部病变存在嗜血管性。(2)部分结节内伴液气平,提示存在液化坏死并与气道相通。(3)存在胸膜下肺气肿。以上3个特点提示病变累及血管和气道。(4)病灶呈现此消彼长的特点,消长可在不同部位出现。从以上影像学特点分析,病因鉴别诊断上应考虑:(1)血管炎:如肉芽肿性多血管炎,可能出现同一时期病灶此消彼长,但患者未经激素治疗,原病灶可完全吸收,且空洞内壁光滑,均与肉芽肿性多血管炎典型表现不符。(2)感染:可出现结节空洞伴出血的感染包括真菌、部分细菌。但真菌一般不出现气液平,且患者无免疫缺陷的宿主因素。细菌感染从临床表现及治疗反应上均不支持。另外,感染性病变较少引起胸膜下肺气肿、气胸等表现,不支持。综上,患者影像学特点用血管炎或感染均难以解释。
张婷(呼吸与危重症医学科):
患者收入我院后再次复查血常规、ESR、C反应蛋白、尿常规均正常。因考虑到肺血管病变,行CT肺动脉造影,结果未发现肺动脉充盈缺损、狭窄闭塞、肺动脉壁增厚或肺动脉畸形等异常表现。遂进行胸腔镜下肺活检。
冯瑞娥(病理科):
患者外院的肺穿刺病理检测仅见出血和坏死表现,未见肺组织,推测穿刺部位为囊腔,诊断意义有限。我院胸腔镜肺活检标本HE染色低倍镜下可见肺组织出血,血肿形成,余肺组织呈轻微肺气肿表现;高倍镜下可见肺泡腔内弥漫性含铁血黄素细胞沉积,局灶出血及血肿形成,血肿周围纤维组织增生,部分纤维组织呈团块状或条索状,伸向肺泡腔,形成机化样改变,但纤维组织较致密,有别于机化性肺炎的疏松结缔组织。另见多发骨化及钙化灶,部分骨化灶位于团块状纤维组织中(图5, 6, 7, 8)。弹力纤维染色未见明确血管弹力纤维断裂。无肉芽肿及血管炎表现。综上,本例病理显示肺出血、机化样纤维化结节及骨化,三种病变同时出现需考虑血管型Ehlers-Danlos综合征(vascular Ehlers-Danlos syndrome,vEDS),建议行基因检测。
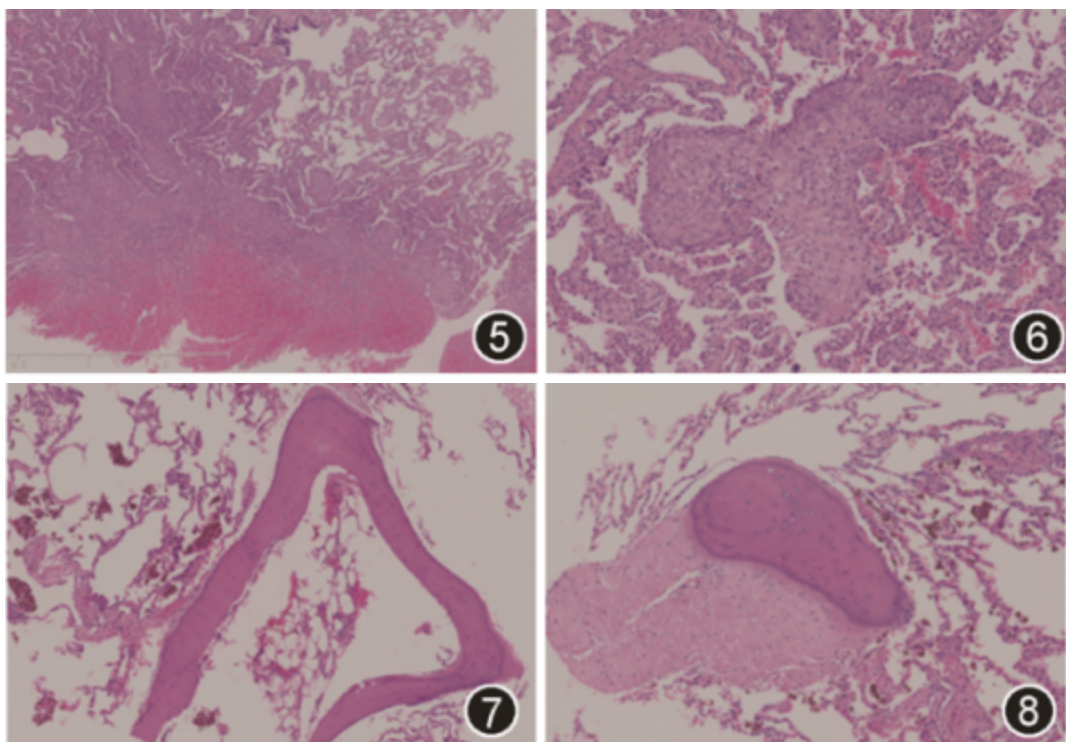
图5 胸腔镜肺活检病理,图片下方见出血及血肿形成,血肿周围纤维组织增生 HE 低倍放大
图6 胸腔镜肺活检病理,可见结节状纤维组织增生,部分伸入到肺泡腔,似机化样纤维化改变 HE 高倍放大
图7 胸腔镜肺活检病理,可见肺组织内骨化,形成板层骨及髓腔,周围肺泡腔内可见灶状吞噬含铁血黄素的巨噬细胞 HE 高倍放大
图8 胸腔镜肺活检病理,纤维性结节内可见骨化 HE 高倍放大
张婷(呼吸与危重症医学科):
采集患者外周血,采用芯片捕获高通量测序进行全外显子组测序检测,发现COL3A1杂合错义突变(c. 2383G>C,p.Gly795Arg)。该突变位于热点突变区域,在ESP、EXAC及千人数据库正常人群中未发现该变异,多种统计方法预测该变异对基因及基因产物造成有害影响。目前尚没有该变异的相关报道,但已有位于同一外显子区域的错义突变(p.Gly786Arg、p.Gly786Val)导致vEDS的文献报道[1, 2, 3]。故判定为疑似致病。综合该患者反复肺出血、自发性气胸、病理表现及基因检测结果,最终诊断为血管型Ehlers-Danlos综合征。
Ehlers-Danlos综合征是一组遗传性结缔组织病,按照2017年国际分类标准分为13型[4],其中血管型(旧称Ⅳ型)是最凶险的一型,约占EDS的5%~10%[5],人群患病率约为1/(50 000~200 000)[6],主要由COL3A1基因突变导致。该病为常染色体显性遗传,但具有阳性家族史者仅约38%[7],约50%的COL3A1突变为新生突变(de novo mutation)[8]。COL3A1编码的Ⅲ型胶原是血管壁、子宫、肠道等内脏器官的主要结构,也存在于肺组织中。当COL3A1基因突变时,会导致Ⅲ型胶原的结构或生成异常,使血管、脏器脆性增加,自发或在较小的应力下出现破裂,导致动脉夹层、动脉破裂、子宫破裂、肠破裂等严重甚至致命性并发症[8]。vEDS的临床特点还包括皮肤松弛、菲薄、小关节过伸及特殊面容[4],但相比经典型EDS程度较轻。胸部病变是vEDS较为少见的并发症,但可先于血管及脏器并发症出现,或作为vEDS的唯一表现[9, 10],本例患者无皮肤松弛或关节过伸表现,暂未出现其他脏器或血管并发症,随访至今,肺部病变是其唯一表现。vEDS的胸部并发症包括自发性气胸、血胸、肺出血、肺气肿等。自发性气胸和血胸是相对多见的胸部并发症,Shalhub等[11]认为年轻患者出现自发性气胸或血胸者应警惕vEDS。肺实质受累以肺出血为主,胸部CT上可出现结节、空洞或囊状影伴周围晕征。病变可自行缩小消退,是其区别于感染、肿瘤的典型特征。少数患者可出现肺内钙化灶,部分患者出现肺气肿或肺大疱表现[12, 13, 14]。
许文兵(呼吸与危重症医学科):
本例患者肺部病理中的出血血肿、异位骨化及纤维性结节等表现是否为vEDS的典型病理表现?如何从病理上解释该患者肺部病灶特点及此消彼长的变化趋势?
冯瑞娥(病理科):
文献报道,vEDS的肺部病变几乎均可见肺出血及血肿形成,多数存在纤维性结节,部分存在骨化[15],本例患者的肺部病理表现与文献报道相符。有研究者推测,肺部影像学的病理机制可能是由于基因突变导致肺组织及肺血管Ⅲ型胶原纤维结构异常,从而易于发生自发性肺撕裂,撕裂累及血管形成出血血肿,或直接因血管破裂导致血肿,随后血肿内出血逐渐吸收形成空腔,周围胶原纤维增生机化形成囊壁,并逐渐向内收缩愈合,最终形成纤维性结节[15, 16]。病理改变过程与影像学上结节、空洞、囊腔的形成及自行吸收缩小的变化趋势相对应。此外,肺泡壁的轻微损伤则可能导致肺气肿或肺大疱。
田欣伦(呼吸与危重症医学科):
冯教授介绍病理中可见骨化,但是患者影像纵隔窗中似乎钙化影并不明显,如何解释?
张婷(呼吸与危重症医学科):
文献报道中,少数患者胸部CT上可出现肺内钙化灶[17, 18],但并非所有病理中存在骨化者CT均可见钙化表现,推测可能与骨化病灶较小,CT分辨率有限相关。
施举红(呼吸与危重症医学科):
EDS是Ⅲ型胶原合成障碍遗传性疾病,是否有好发年龄?诊断标准是否与年龄相关?
张婷(呼吸与危重症医学科):
根据2017年EDS国际分类标准[4],血管型EDS的分类标准为:主要标准:(1)存在COL3A1致病性突变的vEDS家族史。(2)年轻时出现动脉破裂。(3)自发性乙状结肠破裂。(4)无其他原因的孕晚期子宫破裂。(5)非创伤下的颈动脉海绵窦瘘。次要标准:(1)皮肤瘀斑(非创伤导致,面颊/后背等非常见部位)。(2)皮肤薄,皮下静脉可见。(3)特殊面容。(4)自发性气胸。(5)肢端畸形。(6)马蹄内翻足。(7)先天性髋关节脱位。(8)小关节过伸。(9)肌腱及肌肉断裂。(10)圆锥角膜。(11)牙龈萎缩及牙龈脆弱。(12)早发静脉曲张。存在主要标准或多项次要标准,应进一步行vEDS的诊断性检查。分类标准中未提及好发年龄,文献报道中患者绝大多数为青年人,确诊时年龄在30岁左右,约25%患者的首次血管或内脏并发症发生于20岁前,超过80%患者40岁前至少发生过一次血管或内脏并发症[7]。
彭敏(呼吸与危重症医学科):
这类患者应如何治疗?或如何预防血管或脏器破裂的发生?
张婷(呼吸与危重症医学科):
目前vEDS暂无明确可预防血管或脏器并发症的治疗方法。塞利洛尔,一种兼具β1肾上腺素受体阻滞及β2受体激动作用的高血压药物可能有助于减少血管并发症的发生,改善预后[19],但结果仍需进一步证实。目前对于vEDS的治疗建议包括:调整生活方式以减少损伤及血管或脏器破裂的风险;维持血压正常,积极控制高血压;提供个体化应急预案;建立照护团队;若条件允许,在专业医疗机构治疗;每年评估血管(通过多普勒超声、低辐射CTA或MRA)[6]。本例患者至今已随诊3年,血压正常,仍间断少量咯血,尚未出现其他血管或脏器并发症,暂无药物治疗。
施举红(呼吸与危重症医学科):
该患者诊治过程中的经验:(1)患者以肺部受累为唯一表现,缺乏vEDS常见的皮肤、关节特征,也未出现其他血管或脏器并发症,单纯累及肺的vEDS在以往的国内外文献报道均属少见,诊断困难。(2)vEDS属于罕见病,对其临床表现、影像及病理特点,尚缺乏足够认识。本病例是学习vEDS的良好机会:对于以反复咯血、气胸为表现,肺内表现为此消彼长的结节、空洞或囊腔而无明显炎症反应、对抗感染治疗无效的年轻患者,应考虑vEDS。(3)临床怀疑vEDS时,应进一步从皮肤、关节等伴随症状、既往病史、家族史等角度寻找诊断线索,尽早进行基因检测。减少反复有创检查(如反复肺穿刺、外科肺活检等)对患者造成的不必要伤害。(4)外科肺活检并不是所有疾病的诊断金标准。(5)再次强调临床-放射-病理MDT重要性,强调随访的重要性。
参考文献(略)
作者:张婷 罗金梅 彭敏 高莉 田欣伦 许文兵 刘鸿瑞 施举红 冯瑞娥
单位:中国医学科学院北京协和医院呼吸与危重症医学科;北京大学第一医院医学影像科;中国医学科学院北京协和医院病理科
本文转载自订阅号「中华结核和呼吸杂志」
引用本文: 张婷, 罗金梅, 彭敏, 等. 此消彼长的肺内结节空洞 [J] . 中华结核和呼吸杂志, 2022, 45(5) : 475-479. DOI: 10.3760/cma.j.cn112147-20211103-00768.