
 分享
分享
病例简介
患者,男性,20岁。主因“进行性干咳3个月”入院。患者1年前因腹泻诊断为“溃疡性结肠炎”,服用泼尼松45mg/d控制病情。3个月前,患者无明显诱因出现干咳,自行服用抗生素及抗真菌药物,无明显好转。就诊时,查体听诊双肺底可闻及吸气末爆裂音。
辅助检查:
SpO2(不吸氧)为92%。WBC 11.38×109/L,CRP 4.67mg/dL,ESR 37mm/h,血KL-6 3824U/mL(↑),免疫球蛋白、LDH和sACE均正常,RF、ANA、ANCA均未见明显异常,血清沉淀素:曲霉菌抗原2+,血CMV抗原、T-SPOT.TB均阴性。痰病原学阴性。肺功能:FEV1 2.68L,占预计值86.2%,FVC 3.25L,占预计值 72.5%,RV/TLC 25.8%,DLCO占预计值64.5%。胸部CT示双肺弥漫分布边界不清微结节及左肺下叶胸膜下结节影(图1)。

图1 胸部CT
右中叶支气管肺泡灌洗液(BALF)中巨噬细胞比例37%,淋巴细胞比例62%,CD4/CD8 0.84。BALF真菌及分支杆菌培养阴性,TB、NTM及PJP核酸检测阴性。经支气管肺活检(TBLB)示:多发松散非干酪样坏死肉芽肿、淋巴细胞性肺泡炎(图2),符合过敏性肺炎(hypersensitivity pneumonitis, HP)表现。
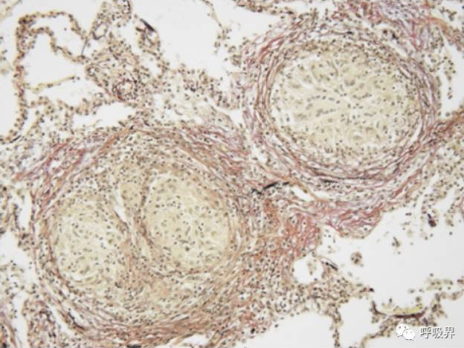
图2 TBLB病理
病例分析
根据以上资料,这例患者应做何诊断?我们再重新梳理一下患者疾病特征:以干咳起病,炎性指标升高,血清沉淀素阳性,肺功能提示限制性通气障碍伴弥散障碍,胸部CT提示双肺弥漫分布边界不清微结节及左肺下叶胸膜下结节,BALF中淋巴细胞比例明显升高,CD4/CD8比例降低,TBLB示多发松散非干酪样坏死肉芽肿、淋巴细胞性肺泡炎。
按照HP的诊断标准:①已知暴露于致敏抗原;②相匹配的临床、影像学或肺功能表现;③BALF淋巴细胞增多;④吸入特定抗原激发试验阳性;⑤组织病理学检查显示松散的非干酪样肉芽肿或单个核细胞浸润。患者满足上述标准中①②③⑤,符合HP诊断。
提出疑问
但是,当我们再次审视患者既往史,不由得产生疑问,一位每天服用中等剂量糖皮质激素的患者,在使用激素过程中出现HP,似乎有些难以解释。那么,这例患者HP样表现的肺部病变,究竟是什么?
因为患者既往有溃疡性结肠炎病史,我们用更加系统的思维再次分析患者的病情:①患者肺部表现可以用溃疡性结肠炎肠外表现解释;②有另一种全身性疾病可以同时引起肠道和肺部病变。
深入分析
1.炎症性肠病(IBD)的肺部表现
虽然IBD肠外表现并不少见,但肺部并发症是其中相对罕见的表现,气道病变、间质性病变、浆膜炎及肺栓塞均可并发于IBD。其中间质性病变主要包括机化性肺炎(OP)、非特异性间质性肺炎(NSIP)、嗜酸性粒细胞性肺炎(EP)和结节病。
上述几种间质性病变中,结节病是唯一以肉芽肿为表现的疾病,但本例患者BALF中CD4/CD8比例降低,并不符合结节病典型表现,且由于患者使用激素过程中IBD无明显活动、结节病理论上对激素存在反应,综合分析本例患者肺部表现不符合IBD肺部受累。
2.同时引起肠道和肺部表现的全身性疾病
常见有结核病、结缔组织病(SLE、SSc、MCTD)、先天性免疫功能障碍和乳糜泻。本例患者结核相关病原及辅助检查均阴性,ANA、ANCA未见明显异常,无慢性腹泻、贫血等表现,故暂不考虑结核病、结缔组织病和乳糜泻。
先天性免疫功能障碍中,起病较晚且以肉芽肿为主要表现的疾病常见有:普通变异型免疫缺陷病(common variable immunodeficiency, CVID)和慢性肉芽肿病(chronic granulomatous disease, CGD)。患者男性,免疫球蛋白正常,更倾向于诊断CGD,尤其需警惕X-连锁慢性肉芽肿病。
求证推断
带着上述疑问,对患者及母亲进行了基因检测,患者gp91phox基因突变阳性,母亲为该基因携带者。故患者最终诊断为X-连锁隐性慢性肉芽肿病,结合临床表现,给予大剂量糖皮质激素治疗后,症状逐渐缓解。复查胸部CT示:微结节较前明显吸收,胸膜下结节较前缩小。
CGD是一种遗传异质性疾病,临床以反复发生的严重细菌及真菌感染和肉芽肿形成为特征,由吞噬细胞中氧化酶(phagocyte oxidase, phox)、即NADPH氧化酶基因缺陷导致。已知致病基因包括gp91phox、p47phox、p22phox、p67phox和p40phox,均为组成NADPH氧化酶复合体的重要基因,其中4个位于常染色体,1个位于X染色体,均为隐性遗传,因此X-连锁CGD更为常见。由于吞噬细胞(中性粒细胞、单核细胞及巨噬细胞)依靠NADPH氧化酶复合体传递电子给分子氧形成超氧化物,行使杀灭病原功能(图3)。因此,这些遗传缺陷会导致吞噬细胞不能有效杀灭微生物,导致患者表现为反复感染;又因为机体杀灭病原能力减弱,病原慢性刺激持续存在而诱发肉芽肿形成,并增加内源性炎症负荷。

图3 CGD发病机制
诊治要点
对疑似CGD的患者,可进行中性粒细胞功能检查进行初筛,确诊则需要基因检查结果。治疗方面:当患者明确存在感染时应积极抗感染,包括细菌及真菌;当患者炎症反应较重时,可使用糖皮质激素和(或)环孢素、硫唑嘌呤等适当给予免疫抑制治疗。
目前异基因造血干细胞移植(allo-HSCT)是CGD患者唯一的治愈手段,基于逆转录病毒的基因治疗仍在积极研究中。
(文/中日医院 王诗尧)